Akromegalie: Definition, Ursachen, Symptome & Behandlung
Akromegalie: Ursachen, Symptome & effektive Behandlungen – früh erkennen, Folgeschäden verhindern. Verständlich erklärt mit Diagnose- und Therapieinfos.
Akromegalie ist ein seltener Hormonzustand, bei dem die vordere Hypophyse (Adenohypophyse) zu viel Wachstumshormon (GH) produziert, nachdem eine Person die Pubertät überschritten hat. Tritt eine Überproduktion von GH vor der Pubertät auf, führt sie hingegen zu Gigantismus. Die häufigste Ursache ist ein gutartiger Tumor der Hypophyse, ein Hypophysenadenom, das aus GH-produzierenden Zellen (Somatotrophen) entsteht. In selteneren Fällen können andere hormonproduzierende Tumoren oder nicht-hypophysäre Ursachen eine Rolle spielen.
Bildergalerie
8 Bilder






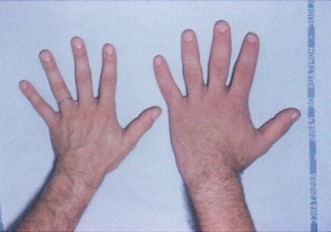
Wie entsteht die Erkrankung (Pathophysiologie)
Das überschüssige Wachstumshormon regt die Leber zur Bildung von Insulin-like Growth Factor 1 (IGF-1) an. IGF-1 vermittelt viele der Wachstumswirkungen von GH und führt zu Vergrößerung von Knochen (insbesondere Hand-, Fuß- und Gesichtsknochen) sowie Weichteilen. Dieser Prozess verläuft langsam, weshalb Veränderungen oft erst Jahre nach Beginn sichtbar werden.
Wer ist betroffen und wie häufig ist Akromegalie?
Akromegalie wird am häufigsten bei Erwachsenen mittleren Alters diagnostiziert (meist zwischen 30 und 50 Jahren), kann aber in jedem Erwachsenenalter auftreten. Die Erkrankung ist selten: die Inzidenz liegt bei wenigen Fällen pro Million Einwohner pro Jahr, die Prävalenz beträgt schätzungsweise mehrere Dutzend Fälle pro Million.
Typische Symptome und Anzeichen
- Vergrößerte Hände und Füße (z. B. plötzlich engere Ringe oder größere Schuhgrößen)
- Veränderungen im Gesicht: vergrößerte Kieferknochen (Prognathie), vergrößerte Nase, hervortretende Stirn (frontal bossing), gröbere Gesichtszüge
- Hautveränderungen: Verdickung, verstärkte Talgproduktion, vermehrtes Schwitzen
- Gelenk- und Wirbelsäulenbeschwerden (Arthralgien, Arthropathie)
- Karpaltunnelsyndrom, Taubheitsgefühle in Händen
- Kopfschmerzen, Sehstörungen (durch Druck des Tumors auf das Sehnervenkreuz, z. B. bitemporale Hemianopsie)
- Atemstörungen im Schlaf (Schlafapnoe)
- Stoffwechselprobleme: Insulinresistenz bis hin zu Diabetes mellitus, erhöhte Blutfettwerte
- Kardiovaskuläre Folgen: Bluthochdruck, Kardiomyopathie
Komplikationen
- Herz-Kreislauf-Erkrankungen (häufigste Ursache erhöhter Mortalität bei unbehandelter Akromegalie)
- Arthropathie und bleibende Gelenkschäden
- Schlafapnoe mit Folgebelastung für Herz und Kreislauf
- Diabetes und metabolisches Syndrom
- Visuelle Einschränkungen bei tumorbedingter Kompression
Diagnostik
- Labor: Bestimmung von IGF-1 (Screeningtest). Ein erhöhtes IGF-1 spricht für eine gesteigerte GH-Wirkung.
- GH-Suppressionstest: oraler Glukosetoleranztest mit Messung der GH-Suppression – bei Akromegalie wird GH nach Glukosegabe nicht ausreichend unterdrückt.
- Bildgebung: Magnetresonanztomographie (MRT) der Hypophyse zur Darstellung eines Adenoms und zur Größen- bzw. Ausbreitungsbeurteilung.
- Weitere Untersuchungen: Hormonprofil der Hypophyse, Visusprüfung und Perimetrie (Gesichtsfeldmessung), kardiometabolische Untersuchung (EKG, Echokardiographie, Blutzucker).
Behandlung
Ziel der Behandlung ist die Normalisierung von GH/IGF-1, Reduktion der Tumormasse und Linderung der Symptome. Therapieentscheidungen erfolgen individuell, meist durch ein interdisziplinäres Team (Endokrinologie, Neurochirurgie, Radiotherapie).
- Operative Entfernung: Transsphenoidale (transnasale) Neurochirurgie ist oft die erste Wahl bei operablen Hypophysenadenomen. Sie kann zu einer schnellen Reduktion des Tumors und der Hormonproduktion führen.
- Medikamentöse Therapie: Somatostatin-Analoga (z. B. Octreotid, Lanreotid) senken GH/IGF-1 und verkleinern manchmal den Tumor; der GH-Rezeptorantagonist Pegvisomant normalisiert IGF-1, wirkt jedoch nicht immer tumorverkleinernd; Dopaminagonisten (z. B. Cabergolin) können bei kleinen Tumoren oder mildem Krankheitsbild wirksam sein.
- Strahlentherapie: Bei unvollständiger Resektion oder unzureichendem Therapieansprechen kann eine stereotaktische Strahlentherapie (z. B. Gamma Knife) oder konventionelle Bestrahlung erwogen werden. Wirkung setzt oft verzögert ein und kann zu Hypopituitarismus führen.
Verlauf, Nachsorge und Prognose
Die Prognose hat sich durch moderne Therapieoptionen deutlich verbessert. Unbehandelt erhöht Akromegalie das Sterberisiko, vor allem durch kardiovaskuläre Erkrankungen. Regelmäßige Nachsorge ist nötig:
- Kontrolle von IGF-1 und GH nach Therapie
- Wiederholtes MRT der Hypophyse zur Verlaufsbeurteilung
- Behandlung und Überwachung von Begleiterkrankungen (Diabetes, Bluthochdruck, Schlafapnoe, Gelenkbeschwerden)
Leben mit Akromegalie
Mit erfolgreicher Therapie können viele Patienten eine deutliche Besserung der Beschwerden und eine Normalisierung der Lebenserwartung erreichen. Neben der medizinischen Behandlung sind physiotherapeutische Maßnahmen, kieferorthopädische bzw. zahnärztliche Betreuung und psychosoziale Unterstützung wichtig. Schwangere Frauen mit Akromegalie sollten engmaschig von einem spezialisierten Team betreut werden.
Da die Veränderungen oft schleichend beginnen, liegt die Diagnose meist erst 8–12 Jahre nach Auftreten der ersten Symptome. Bei Verdacht auf Akromegalie ist eine frühzeitige Abklärung durch einen Endokrinologen ratsam, um Komplikationen zu vermeiden oder zu begrenzen.
Anzeichen und Symptome
- Kopfschmerzen
- Sehprobleme
- Hirsutismus (bei Frauen): das Wachstum von Haaren im Gesicht.
- Stirnvorwölbung: Der Knochen unterhalb der Stirn wird größer als normal.
- Prognose: Ein Teil des Gesichts ragt mehr als normal heraus, in der Regel der Unterkiefer.
- Müdigkeit: Die Person ist oft müde.
- Hoher Blutdruck
- Skin-Tags: Kleiner warzenartiger Wuchs auf der Haut, der nicht krebsartig ist.
- Hyperhidrose: wenn eine Person zu stark schwitzt.
- Bromhidrose: Körpergeruch
- Hepatomegalie
- Vergrößerung von Händen und Füßen
- Kardiomyopathie
- Dickdarmkrebs
Fragen und Antworten
F: Was ist Akromegalie?
A: Akromegalie ist ein medizinischer Zustand, der auftritt, wenn die Hypophyse nach der Pubertät zu viel Wachstumshormon produziert.
F: Was verursacht Gigantismus?
A: Gigantismus wird durch eine übermäßige Menge an Wachstumshormonen verursacht, die von der Hypophyse vor der Pubertät produziert werden.
F: Was ist die häufigste Ursache für Akromegalie?
A: Die häufigste Ursache für Akromegalie ist das Vorhandensein eines Hypophysenadenoms, eines Tumors an der Hirnanhangsdrüse.
F: Wer ist am meisten gefährdet, an Akromegalie zu erkranken?
A: Akromegalie wird am häufigsten bei Erwachsenen im mittleren Alter diagnostiziert.
F: Was sind die möglichen Folgen einer unbehandelten Akromegalie?
A: Unbehandelte Akromegalie kann zu schweren Entstellungen, ernsten Komplikationen und frühem Tod führen.
F: Warum ist Akromegalie schwer zu diagnostizieren?
A: Akromegalie ist schwer zu diagnostizieren, wenn sie gerade erst begonnen hat, und wird in der Regel erst 10-12 Jahre nach Beginn der Erkrankung entdeckt.
F: Was sind die sichtbaren Symptome der Akromegalie?
A: Das auffälligste Symptom der Akromegalie ist die Veränderung des Aussehens, insbesondere des Gesichts.
Verwandte Artikel
Autor
AlegsaOnline.com Akromegalie: Definition, Ursachen, Symptome & Behandlung Leandro Alegsa
URL: https://de.alegsaonline.com/art/762