Computergestützte Chemie: Definition, Methoden und Anwendungen
Computergestützte Chemie: Methoden, Anwendungen & Vorhersagen — von Molekülstrukturen über Reaktivität bis Medikamenten- und Materialentwicklung. Theorie, Simulationen & Praxiseinsatz.
Die computergestützte Chemie ist ein Zweig der Chemie, der die Informatik zur Lösung chemischer Probleme einsetzt. Diese Programme berechnen die Strukturen und Eigenschaften von Molekülen und Festkörpern. Die computergestützte Chemie ergänzt normalerweise die durch chemische Experimente gewonnenen Informationen. Sie kann chemische Phänomene vorhersagen, die noch nicht beobachtet worden sind. Sie ist bei der Entwicklung neuer Medikamente und Materialien weit verbreitet.
Bildergalerie
8 Bilder


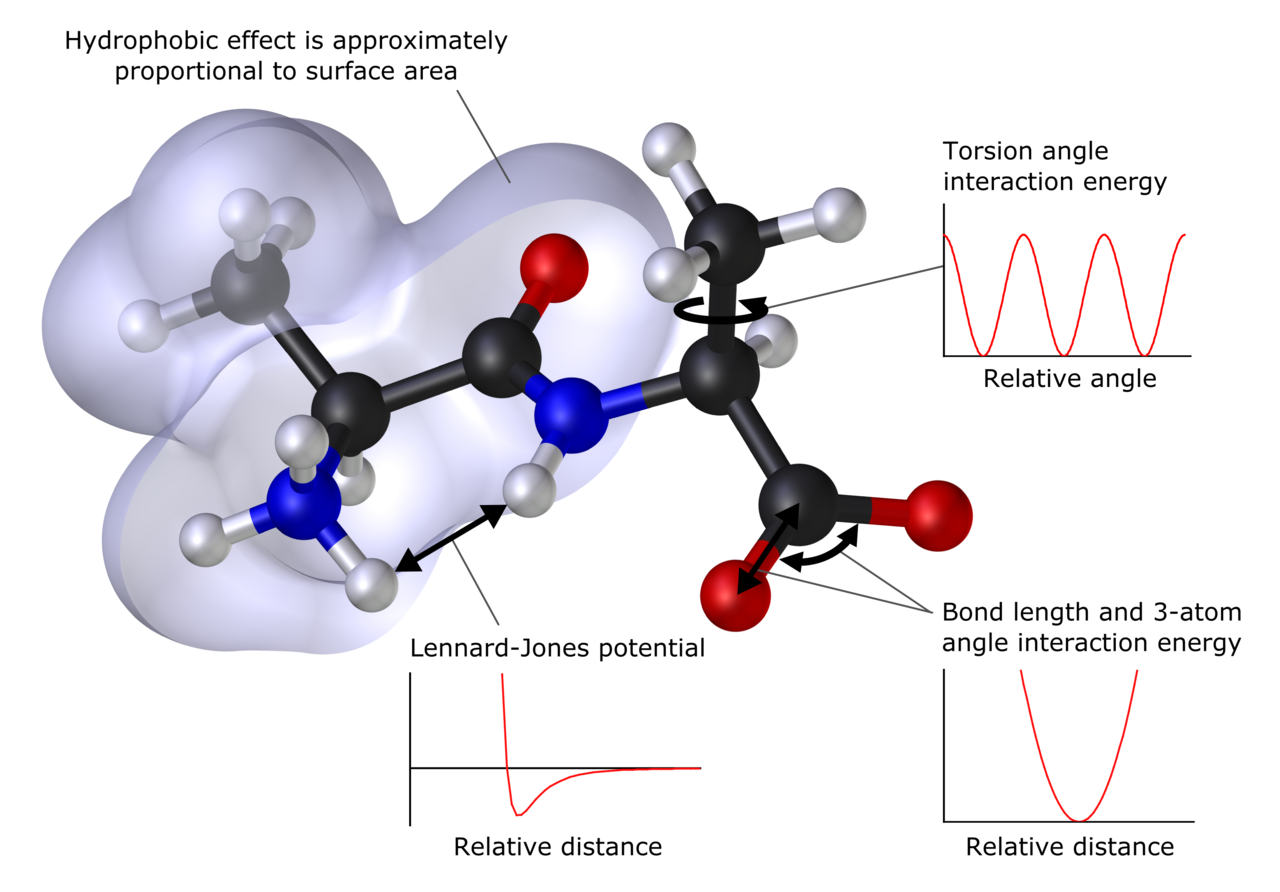


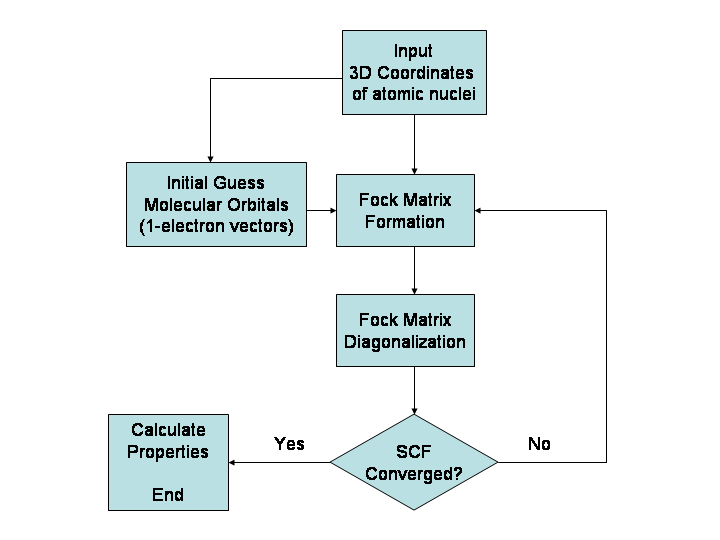
Was kann vorhergesagt werden
Die computergestützte Chemie kann Struktur (d.h. die erwarteten Positionen der Atome des Moleküls), absolute und relative (Wechselwirkungs-)Energien, elektronische Ladungsverteilungen, Dipole und höhere Multipolmomente, Schwingungsfrequenzen, Reaktivität oder andere spektroskopische Größen sowie Querschnitte für Kollisionen mit anderen Teilchen vorhersagen.
Darüber hinaus lassen sich mit geeigneten Methoden auch Übergangszustände und Reaktionswege (Potentialflächen), Aktivierungsenergien, thermodynamische Größen (Enthalpien, freie Energien, Entropien), elektronische Anregungszustände (z. B. UV/Vis-Absorption mittels TD‑DFT) sowie kinetische Parameter berechnen. Mit statistischen Sampling‑Verfahren und molekulardynamischen Simulationen können Temperatur-, Druck‑ und Lösungsmittel‑Effekte sowie Transport‑ und Diffusionsprozesse modelliert werden.
Methoden und ihre Einordnung
Die computergestützte Chemie betrachtet sowohl statische als auch dynamische Systeme. In allen Fällen nehmen mit der Größe des untersuchten Systems auch die Rechenzeit und andere Ressourcen (wie Speicher und Plattenplatz) zu. Dieses System kann ein einzelnes Molekül, eine Gruppe von Molekülen oder ein Feststoff sein. Die Methoden der computergestützten Chemie reichen von hochgenau bis sehr approximativ. Hochgenaue Methoden sind in der Regel nur für kleine Systeme durchführbar.
- Ab initio (quantentheoretische) Methoden: Dazu zählen Hartree‑Fock und darauf aufbauende korrelierte Methoden wie MP2, CCSD(T) oder multireferenzielle Verfahren (CASSCF, CASPT2). Sie liefern sehr genaue Ergebnisse für kleine bis mittlere Systeme, sind aber rechenintensiv (Skalierung oft zwischen N3 und N7, wobei N die Zahl der Basisfunktionen ist).
- Dichtefunktionaltheorie (DFT): Weit verbreitet wegen des günstigen Kompromisses zwischen Genauigkeit und Kosten. Wichtig sind die Wahl des Funktionals (z. B. GGA, Hybrid, meta‑GGA) und des Basissatzes. DFT eignet sich gut zur Struktur‑ und Energieabschätzung, zur Modellierung von Festkörpern (mit planewave‑Ansatz) und zur Beschreibung vieler Reaktionsprozesse.
- Semiempirische Methoden: Reduzieren Rechenaufwand durch parametrische Vereinfachungen (z. B. PMx, AM1). Geeignet für sehr große Systeme oder schnelle Screening‑Berechnungen, mit geringerer Genauigkeit.
- Molekulare Mechanik (MM) und Kraftfeld‑Methoden: Beschreiben Atome als Punktmassen mit klassischen Potentialen (Bond, Winkel, Torsion, van der Waals, Coulomb). Ermöglichen Simulationen tausender bis Millionen von Atomen (Kraftfelder: AMBER, CHARMM, OPLS, GROMOS u. a.).
- Molekulardynamik (MD) und Monte‑Carlo (MC): Dynamische Simulationen zur Untersuchung zeitabhängiger Phänomene, Konformationsräume, Diffusion, Temperatur‑ und Lösungseffekte. MD kann mit klassischen Kraftfeldern oder als ab initio MD (z. B. Car–Parrinello, Born‑Oppenheimer) durchgeführt werden.
- QM/MM‑Methoden: Hybridansatz, bei dem ein kleiner relevanter Bereich (Reaktionszentrum) quantenmechanisch und die Umgebung klassisch behandelt wird — nützlich für Enzyme, Katalyse und große Systeme.
- Spezialmethoden: Zeitskalenüberschreitende Methoden (Enhanced sampling, metadynamics), Solvatationsmodelle (implizit: PCM/COSMO; explizit: mit Lösungsmittelmolekülen), und Methoden für angeregte Zustände (TD‑DFT, EOM‑CC, CI).
- Maschinelles Lernen und Datengetriebene Modelle: ML‑Potentiale (Neural Network Potentials, Gaussian Approximation Potentials), Property‑Prediction‑Modelle und Beschleuniger für Screening‑Workflows gewinnen stark an Bedeutung.
Typischer Arbeitsablauf (Workflow)
- Modellaufbau: Geometrieerstellung, Protonierung, Konformationsanalyse.
- Geometrieoptimierung: Finden lokaler Minima auf der Potentialfläche.
- Frequenzberechnung: Bestätigung von Minima vs. Übergangszuständen und Berechnung thermodynamischer Korrekturen.
- Eigenschaftsberechnung: Energien, Dipole, spektroskopische Signale, Reaktionsprofile.
- Validierung: Vergleich mit experimentellen Daten oder höheren Rechnungsstufen.
- Sampling/Dynamik: MD‑Simulationen oder Monte‑Carlo zur Untersuchung von Temperatur‑ und Zeitabhängigkeiten.
- High‑Throughput und Screening: Automatisierte Berechnungen großer Datenmengen zur Identifikation vielversprechender Kandidaten.
Anwendungen
- Wirkstoffdesign: Ligandenoptimierung, Bindungsenergien, virtuelle Screening‑Bibliotheken, Vorhersage von ADME‑Eigenschaften.
- Materialwissenschaften: Design von Halbleitern, Katalysatoren, Polymermaterialien, Batteriematerialien und Festkörperphasen.
- Katalyse und Reaktionsmechanismen: Aufklärung von Reaktionspfaden, Identifikation von Übergangszuständen und Energetik.
- Spektroskopie: Simulation und Interpretation von IR, Raman, NMR, UV/Vis und Röntgenspektren.
- Umwelt‑ und Atmosphärenchemie: Reaktionsmechanismen von Schadstoffen, Photochemie und Kinetik.
- Astro‑ und physikalische Chemie: Modellierung interstellarer Moleküle, Kollisionen und Plasmaprozesse.
- Industrielle Prozesse: Optimierung von Synthesewegen, Materialtests und Scale‑up‑Vorhersagen.
Stärken und Grenzen
Computergestützte Chemie bietet tiefe Einsichten in Struktur‑Eigenschafts‑Beziehungen und erlaubt Vorhersagen, die Experimente leiten und Kosten senken. Einschränkungen ergeben sich durch Modellannahmen (z. B. Vernachlässigung von Elektronenkorrelation, Einsatz approximativer Funktionale oder Kraftfelder), Rechenkosten und die Notwendigkeit sorgfältiger Validierung. Für große Systeme sind oft hybride oder approximative Verfahren notwendig, und Ergebnisse sollten immer im Kontext experimenteller Daten bewertet werden.
Software, Hardware und Trends
Es gibt eine Vielzahl von Softwarepaketen für unterschiedliche Aufgabenbereiche (quantum chemistry, DFT‑Codes, MD‑Engines, Multiskalen‑Pakete). Moderne Entwicklungen nutzen Hochleistungsrechner, GPUs und verteiltes Rechnen; außerdem spielen Automatisierung, Workflow‑Management und Integration von maschinellem Lernen eine wachsende Rolle. Ziele der Forschung sind höhere Genauigkeit bei geringeren Kosten, bessere Skalierbarkeit für große Systeme und engere Verzahnung mit experimentellen Methoden.
Insgesamt ist die computergestützte Chemie ein vielseitiges und sich schnell entwickelndes Feld, das von Grundlagenforschung bis zur industriellen Anwendung reicht und zunehmend durch datengetriebene Ansätze ergänzt wird.

Verwandte Seiten
- Bioinformatik
- Statistische Mechanik
Fragen und Antworten
F: Was ist computergestützte Chemie?
A: Die computergestützte Chemie ist ein Zweig der Chemie, der die Computerwissenschaft zur Lösung chemischer Probleme einsetzt. Sie kann dazu verwendet werden, die Strukturen und Eigenschaften von Molekülen und Festkörpern zu berechnen, chemische Phänomene vorherzusagen, die noch nicht beobachtet wurden, und neue Medikamente und Materialien zu entwickeln.
F: Welche Arten von Systemen werden in der Computerchemie untersucht?
A: Die Computerchemie befasst sich sowohl mit statischen als auch mit dynamischen Systemen. Das System kann ein einzelnes Molekül, eine Gruppe von Molekülen oder ein Festkörper sein.
F: Welche Arten von Informationen kann die computergestützte Chemie liefern?
A: Die computergestützte Chemie kann Informationen wie die Struktur (Positionen der Atome), absolute und relative Energien, elektronische Ladungsverteilungen, Dipole und höhere Multipolmomente, Schwingungsfrequenzen, Reaktivität oder andere spektroskopische Größen sowie Wirkungsquerschnitte für Kollisionen mit anderen Teilchen liefern.
F: Wie genau sind die in der computergestützten Chemie verwendeten Methoden?
A: Die Genauigkeit der in der computergestützten Chemie verwendeten Methoden reicht von sehr genau bis hin zu sehr grob. Hochpräzise Methoden sind in der Regel nur für kleine Systeme anwendbar.
F: Wie ergänzt die computergestützte Chemie die experimentellen Daten?
A: Die computergestützte Chemie ergänzt normalerweise die durch chemische Experimente gewonnenen Informationen. Sie kann verwendet werden, um Ergebnisse vorherzusagen, die noch nicht experimentell beobachtet worden sind.
F: Beeinflusst die Größe des zu untersuchenden Systems, wie viel Computerzeit benötigt wird?
A: Ja - je größer das untersuchte System ist, desto mehr Computerzeit wird für die Analyse benötigt und desto mehr Ressourcen wie Arbeitsspeicher und Festplattenplatz werden für die Speicherung benötigt.
Verwandte Artikel
Autor
AlegsaOnline.com Computergestützte Chemie: Definition, Methoden und Anwendungen Leandro Alegsa
URL: https://de.alegsaonline.com/art/22297