Thalassämie: Ursachen, Symptome und Behandlung der genetischen Blutkrankheit
Thalassämie: Ursachen, Symptome & Behandlung der genetischen Blutkrankheit – Ursachen, Diagnose, Therapie (Transfusion, Eisenchelation, Stammzelltransplantation) und Prognose.
Thalassämie (auch als Thalassämie bezeichnet) ist eine genetische Erkrankung des Blutes, die ursprünglich im Mittelmeerraum beschrieben wurde, heute aber in vielen Regionen der Welt verbreitet ist.
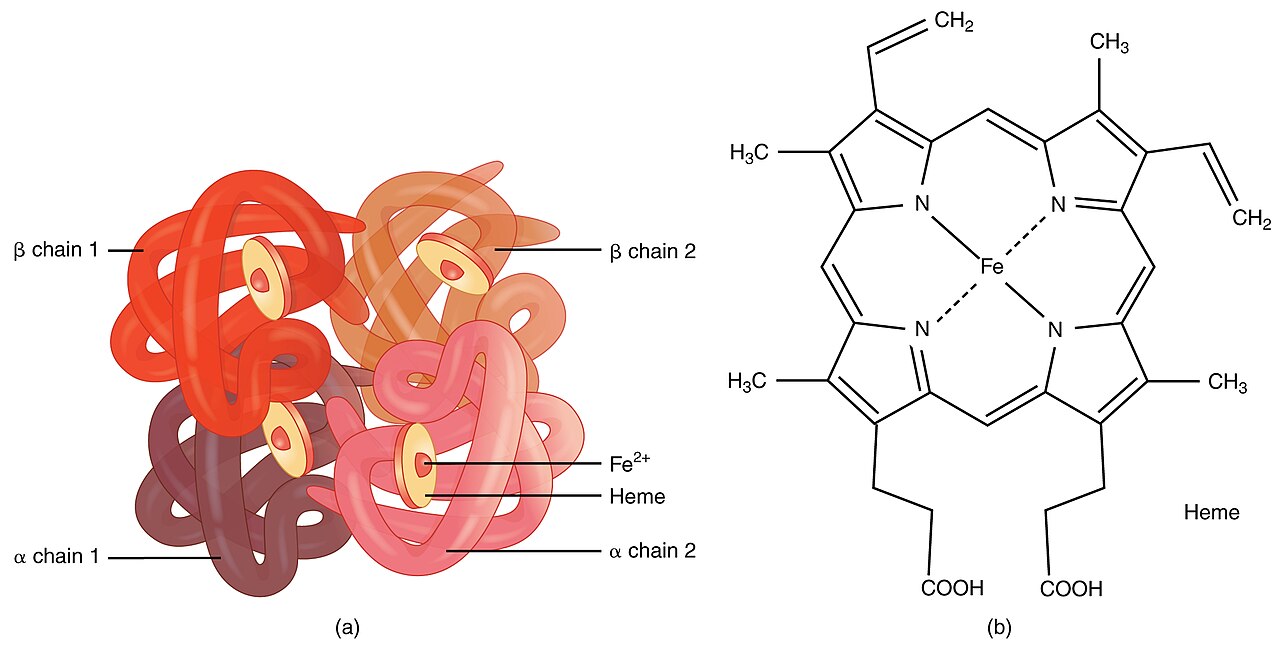
Die Krankheit entsteht durch eine Störung in der Produktion von Hämoglobin: Mutationen in bestimmten Genen führen dazu, dass der Körper weniger oder fehlerhaftes Hämoglobin produziert. In der Folge werden rote Blutkörperchen geschwächt und häufig vorzeitig zerstört. Menschen mit Thalassämie haben deshalb weniger zirkulierende rote Blutkörperchen und weniger funktionsfähiges Hämoglobin, was zu einer leichten bis schweren Anämie führt.
Bildergalerie
8 Bilder





Ursachen und Vererbung
Thalassämie wird durch Veränderungen (Mutationen oder Deletionen) in Genen verursacht, die für die Hämoglobin-Untereinheiten verantwortlich sind. Man unterscheidet vor allem zwei Haupttypen:
- Alpha-Thalassämie: betrifft die Gene HBA1 und HBA2 (meist durch Deletionen); Schweregrad hängt von der Zahl der betroffenen Allele ab.
- Beta-Thalassämie: wird meist durch Punktmutationen im HBB-Gen verursacht; auch hier reicht das Spektrum von asymptomatischen Trägern bis zur schweren Form (Thalassämia major).
Die Erkrankung wird in der Regel autosomal rezessiv vererbt. Träger sind heterozygot für das Thalassämie-Allel (nur eines der beiden Allele ist mutiert). Wenn zwei Träger ein Kind bekommen, besteht ein erhöhtes Risiko für eine schwere Form.
Warum die Mutation weiterbesteht
In Regionen mit Malariarisiko bietet das Trägerdasein einen gewissen Schutz vor Malaria. Dieser sogenannte heterozygote Vorteil erklärt, warum die Thalassämie-Mutation in manchen Populationen häufiger vorkommt als allein aufgrund von Neumutationen zu erwarten wäre. In diesem Zusammenhang ähnelt die Thalassämie anderen Hämoglobin-Störungen wie der Sichelzellkrankheit.
Symptome und Komplikationen
Die Beschwerden hängen vom Typ und Schweregrad ab:
- Leichte Formen: meist kaum Symptome, gelegentliche Müdigkeit, Blässe.
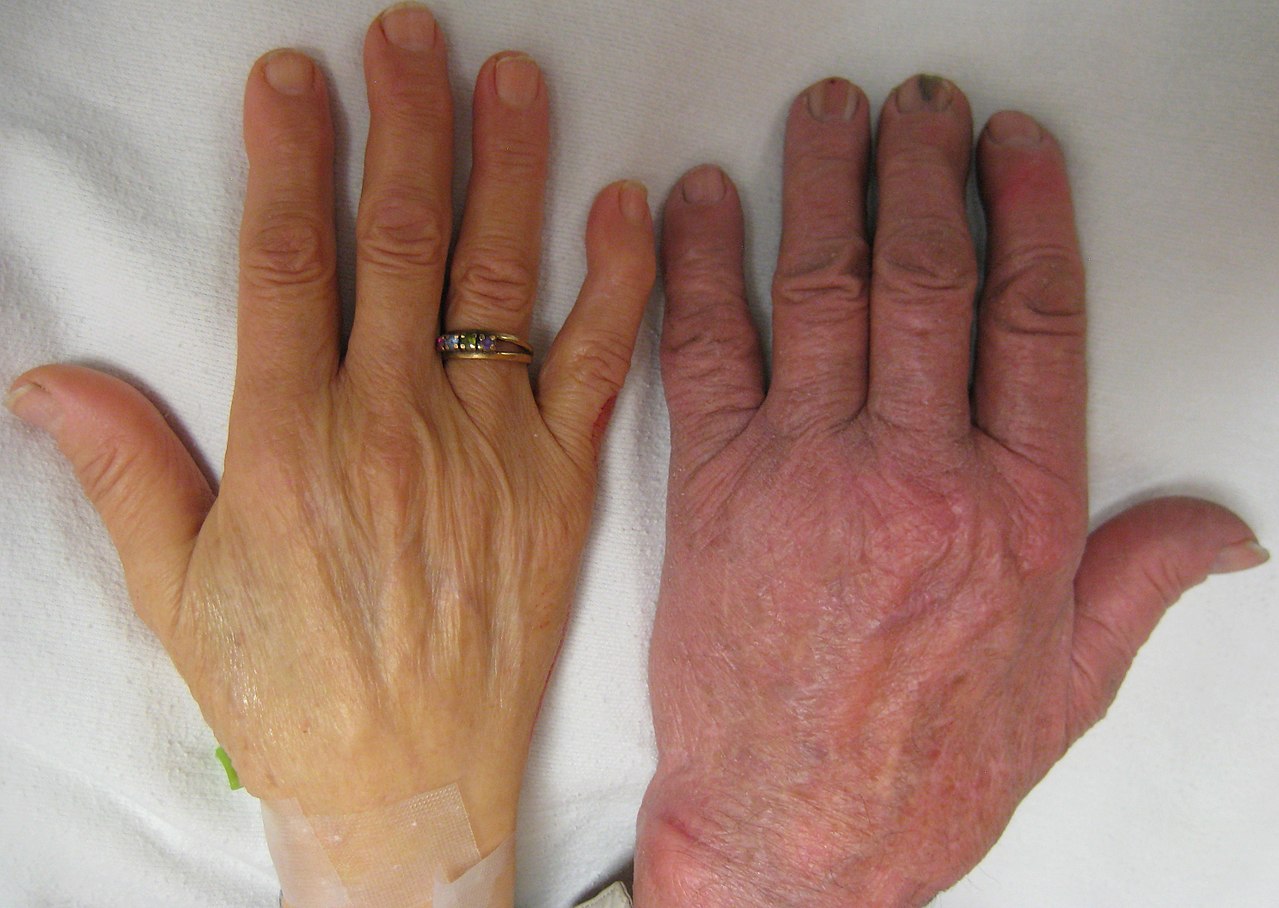
- Mittelschwere bis schwere Formen: ausgeprägte Müdigkeit, Atemnot bei Belastung, Blässe, Gelbsucht (durch erhöhten Hämoglobinabbau), vergrößerte Milz (Splenomegalie), verlangsamtes Wachstum bei Kindern und Entwicklungsverzögerungen.
Mögliche Komplikationen umfassen:
- Eisenüberladung (meist Folge regelmäßiger Bluttransfusionen und erhöhter Eisenaufnahme), die Leber, Herz und endokrine Organe schädigen kann.
- Knochenveränderungen und Deformationen durch gesteigerte Blutbildungsaktivität im Knochenmark.
- Erhöhte Infektionsanfälligkeit und bei schwerer Erkrankung Risiko für schwere Infektionen wie Lungenentzündung.
- Herz-Kreislauf-Erkrankungen bis hin zur Herzinsuffizienz.
Diagnose
Typische diagnostische Schritte:
- Blutbild (CBC): zeigt oft mikrozytäre, hypochrome Anämie mit erniedrigter Hämoglobinkonzentration und niedrigem MCV.
- Peripheres Blutausstrich: kann Zielzellen und andere charakteristische Veränderungen zeigen.
- Hämoglobin-Analysen (z. B. Hämoglobin-Elektrophorese, HPLC): zeigen bei Beta-Thalassämie charakteristische Veränderungen (z. B. erhöhtes HbA2, HbF).
- Retikulozytenzahl, Serumferritin und andere Tests zur Abklärung einer Eisenüberladung oder Eisenmangel.
- Genetische Tests: molekulargenetische Untersuchungen bestätigen die Mutation und sind wichtig für Beratung und pränatale Diagnostik.
Behandlung
Die Therapie richtet sich nach Schweregrad und Symptomen:
- Beobachtung und symptomatische Behandlung bei milden Formen (Trägerzustand).
- Regelmäßige Bluttransfusionen bei schwerer Beta-Thalassämie (Thalassämia major), um eine ausreichende Hämoglobinversorgung sicherzustellen.
- Eisenchelation zur Vermeidung und Behandlung der Eisenüberladung (z. B. Deferoxamin, Deferasirox, Deferipron).
- Operative Maßnahmen wie Splenektomie in ausgewählten Fällen (z. B. wiederholte Transfusionsbedürftigkeit durch vergrößerte Milz).
- Allogene Stammzell- bzw. Knochenmarktransplantation kann bei passenden Spendern eine Heilung ermöglichen; Voraussetzung ist ein HLA-kompatibler Spender (z. B. Geschwister). Die Behandlung birgt Risiken und erfordert spezialisierte Zentren.
- Neue Therapieoptionen: Gentherapie-Ansätze und gentherapeutische Korrekturen (z. B. gentherapeutische Gentransfer- oder Geneditierungstechniken) zeigen vielversprechende Ergebnisse und sind teils bereits zugelassen bzw. in Studien.
- Zusätzliche Maßnahmen: Impfschutz, Behandlung von Infektionen, Ernährungsberatung, Osteoporose-Prophylaxe und endokrinologische Kontrolle bei Hormonstörungen.
Prävention und genetische Beratung
In betroffenen Regionen werden Screening-Programme für werdende Eltern angeboten. Maßnahmen:
- Trägerdiagnostik (Bluttests, Hämoglobin-Analysen, genetische Tests).
- Genetische Beratung für Paare mit Risiko, damit sie die Chancen und Optionen (pränatale Diagnostik, Präimplantationsdiagnostik) verstehen.
- Aufklärung und Vorsorgeuntersuchungen in Regionen mit hoher Prävalenz.
Prognose
Mit moderner Therapie und guter interdisziplinärer Betreuung können viele Betroffene ein wesentlich verbessertes Leben erreichen; schwere Fälle benötigen jedoch lebenslange Versorgung. Gute Therapie kann Komplikationen wie Herz- oder Lebererkrankungen verzögern oder mindern, während unbehandelten schweren Formen eine deutlich schlechtere Prognose droht.
Verbreitung
Thalassämie ist besonders häufig im Mittelmeerraum, im Nahen Osten, in Teilen Afrikas, Südasien und Südostasien. Historisch hat der Schutz gegen Malaria zur Häufung der Trägergene in diesen Regionen beigetragen.
Zusammenfassend ist Thalassämie eine genetisch bedingte Störung der Hämoglobinproduktion mit sehr unterschiedlichen Erscheinungsformen. Früherkennung, gezielte Therapie (inkl. Transfusionen und Eisenchelation) sowie genetische Beratung sind entscheidend für die Lebensqualität und Prognose der Betroffenen.
Fragen und Antworten
F: Was ist Thalassaemie?
A: Thalassämie ist eine genetische Störung des Blutes, die ursprünglich aus dem Mittelmeerraum stammt. Diese Krankheit wird durch die Schwächung und Zerstörung der roten Blutkörperchen aufgrund mutierter Gene verursacht, die die Hämoglobinbildung im Körper beeinflussen.
F: Welche Komplikationen sind mit Thalassämie verbunden?
A: Zu den Komplikationen im Zusammenhang mit Thalassämie können Lungenentzündung, Eisenüberladung, Knochendeformationen und Herz-Kreislauf-Erkrankungen gehören.
F: Wie schützt die Thalassämie vor Malaria?
A: Träger der Thalassämie haben einen selektiven Überlebensvorteil für Träger (bekannt als heterozygoter Vorteil), der dazu beiträgt, dass die Mutation in Populationen weit über der Mutationsrate liegt. Dadurch sind sie vor Malaria geschützt, die in Regionen, in denen dieses Merkmal weit verbreitet ist oder war, häufig vorkommt.
F: Gibt es verschiedene Versionen der Thalassämie?
A: Ja, es gibt eine Reihe verschiedener Versionen der Thalassämie, die jeweils durch eine Mutation an einer anderen Stelle im Genom verursacht werden. Sie ähnelt einer anderen genetischen Störung, die das Hämoglobin betrifft, der Sichelzellenkrankheit.
F: Ist es möglich, Patienten mit Thalassämie zu heilen?
A: Ja, es ist möglich, Patienten mit Thalassämie durch Knochenmarktransplantationen von kompatiblen Spendern zu heilen, die einen HLA-kompatiblen Spender haben.
Verwandte Artikel
Autor
AlegsaOnline.com Thalassämie: Ursachen, Symptome und Behandlung der genetischen Blutkrankheit Leandro Alegsa
URL: https://de.alegsaonline.com/art/97356
Quellen
- accessmedicine.com : accessmedicine.com/content.aspx?aID=6123722
- mayoclinic.com : mayoclinic.com/health/thalassemia/DS00905/DSECTION=complications
- bloodjournal.hematologylibrary.org : HLA-matched sibling bone marrow transplantation for β-thalassemia major