Sichelzellenanämie
Die Sichelzellenanämie ist eine genetische Erkrankung. Sie befällt rote Blutkörperchen. Sie verwandelt die Zellen von flexiblen Scheiben in starre Halbmonde. Wenn viele rote Blutkörperchen diese Form annehmen, verstopfen die Venen. Dies kann zu…
Die Sichelzellenanämie ist eine genetische Erkrankung. Sie befällt rote Blutkörperchen. Sie verwandelt die Zellen von flexiblen Scheiben in starre Halbmonde. Wenn viele rote Blutkörperchen diese Form annehmen, verstopfen die Venen. Dies kann zu Schäden an vielen Organen führen. Die Organschäden nehmen mit der Zeit zu und führen zu einem frühen Tod.
Bildergalerie
8 Bilder






Die Krankheit
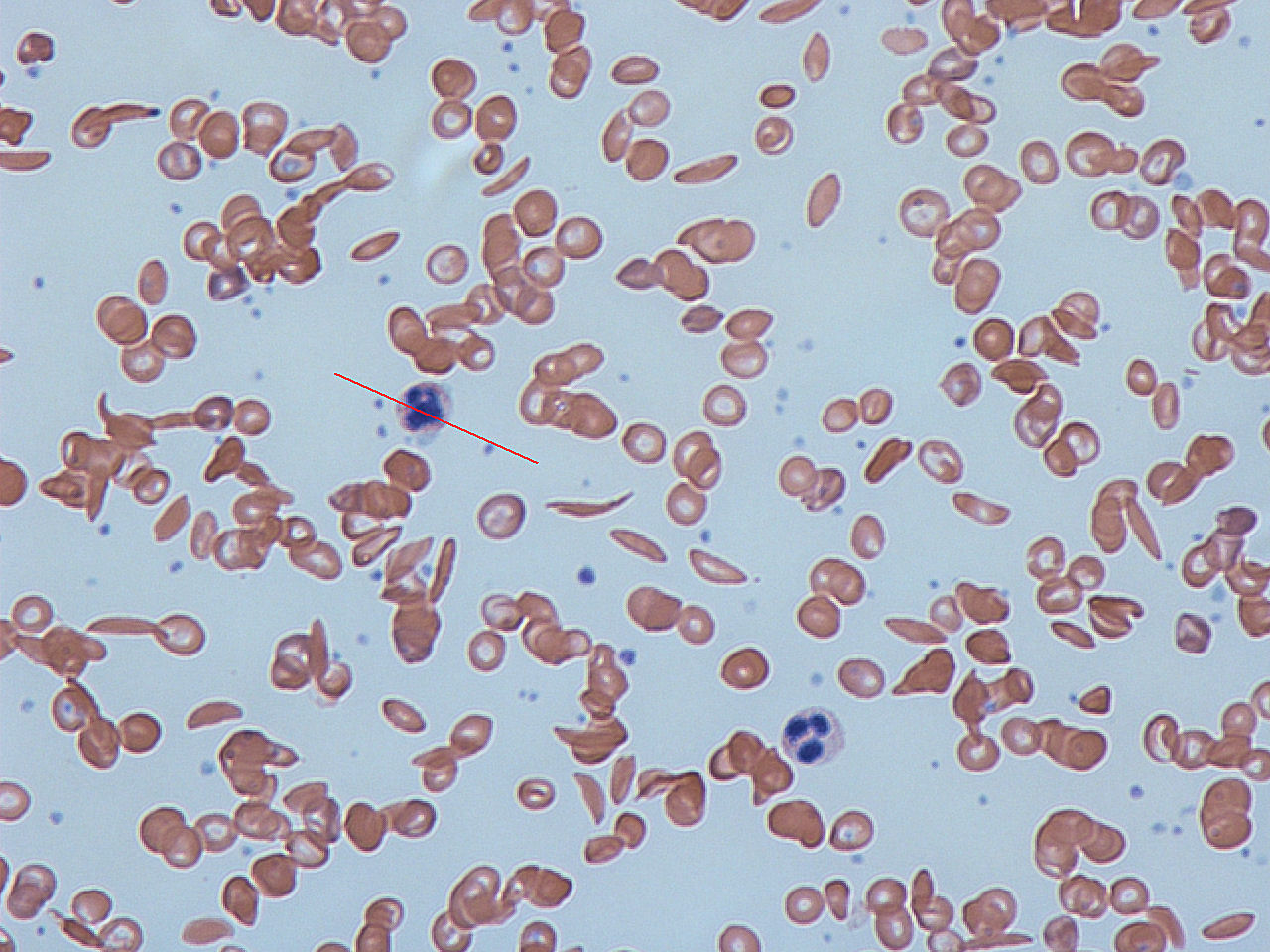
Dies ist eine lebenslange Krankheit, die in der Kindheit beginnt. Die roten Blutkörperchen nehmen eine abnorme, starre, sichelförmige Gestalt an. Die Zellen werden auch klebrig. Dadurch wird der Blutfluss erschwert, wenn die Zellen durch lange, enge Kapillaren fließen. Sauerstoffmangel verstärkt das Problem. Wenn sie durch sauerstoffarme Bereiche strömen, nehmen die meisten Zellen diese Form an. Die Zellen kleben dann an der Innenwand von Blutgefäßen, insbesondere an der Verzweigungsstelle von Venen. Dies führt zu einer Blockade des Blutflusses in vielen Organen. Schwere Komplikationen können die Folge sein.
Das klassische Beispiel für eine Sichelzellkrise ist das "Akute Brustsyndrom" (ACS). Dieses Syndrom tritt nur bei Sichelzellen auf und kann ohne Behandlung innerhalb von ein oder zwei Tagen zum Tod führen. In der Vergangenheit wurde das akute Brustsyndrom anders betrachtet als eine Infektion (Lungenentzündung). Bei der Behandlung macht es jedoch nicht viel Sinn, diese Unterscheidung zu treffen.
ACS ist eine klinische Diagnose, die durch mindestens eine Thorax-Röntgenaufnahme unterstützt wird. In allen anderen Organen verursacht Sauerstoffmangel eine Erweiterung der Blutgefäße. Aber die Lunge ist ein einzigartiges Organ, in dem sich die Blutgefäße bei Sauerstoffmangel verengen. Dieses einzigartige Problem macht die Lunge zu einem Hauptziel der Krankheit. Fieber ist das häufigste Symptom der ACS bei Kindern, da Infektionen häufiger auftreten. Bei Erwachsenen können zirkulierende Blutgerinnsel und Knochenmarksplitter ebenfalls zur Verstopfung von Gefäßen in der Lunge beitragen und zu ACS führen. ACS kann teilweise durch Bluttransfusionen behandelt werden, um die sichelförmigen Zellen mit einigen normalen roten Blutkörperchen zu verdünnen. Eine noch bessere Behandlung ist ein Verfahren, das als Erythrozytenaustausch bezeichnet wird. Automatisierte Apheresegeräte können den Erythrozytenaustausch durchführen.
Ein milderes und häufigeres Problem ist die "schmerzhafte Krise". Zu einer schmerzhaften Krise gehören Flanken-, Rücken- und Oberschenkelschmerzen, die durch eine Behandlung gelindert werden können. Eine schmerzhafte Krise kann sich zu schlimmeren Problemen wie akutem Brust- und anderen Organversagen, z.B. Schlaganfall, Herzinfarkt, entwickeln. Sowohl Schlaganfall als auch Herzinfarkt sind allgemeine Probleme, die bei älteren Menschen auftreten können. Bei Sichelpatienten können diese aber auch schon in jungen Jahren auftreten.
Die Milz ist bei verschiedenen ethnischen Gruppen mit dieser Erkrankung unterschiedlich betroffen. Die Milz ist das Organ, das alte Erythrozyten filtert und zerstört. Alte Erythrozyten sind steif und können nicht durch einige sehr enge Schlitze in der Milz passieren. Aber bei Sichelpatienten werden alle Zellen sehr schnell steif und verstopfen so die Milz weiter. Schon ab einem sehr jungen Alter sterben Teile der Milz an diesem Problem ab. Bei der reinen Form dieser Erkrankung ist die gesamte Milz abgestorben und geschrumpft, bevor der Patient erwachsen wird. In der normalen Milz befindet sich ein großer Vorrat an B-Zellen, die Antikörper bilden und uns vor Bakterien schützen. Der Verlust einer funktionierenden Milz führt zum Verlust des Schutzes vor solchen Bakterien.
In vielen asiatischen Populationen tritt die Beta-Thalassämie zusammen mit der Sichelzellkrankheit auf. Die Thalassämie selbst ist eine weitere Form der Anämie. Die Natur der beiden Krankheiten ist jedoch entgegengesetzt. Die Thalassämie erhöht die Erythrozytenflexibilität. Die Thalassämie selbst kann jedoch eine ernsthafte Erkrankung sein.
Populationsgenetik
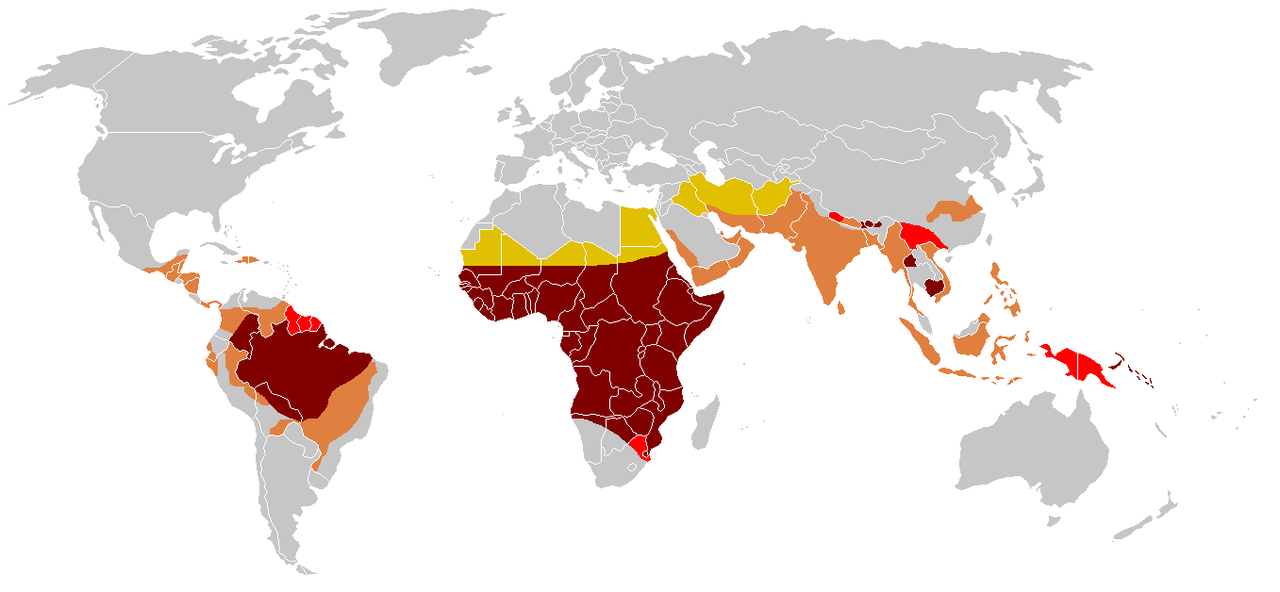
Die Sichelbildung erfolgt aufgrund einer einzigen Punktmutation im Gen für die Beta-Kette des Hämoglobins. Die Sichelzellkrankheit tritt häufiger bei Menschen (oder deren Nachkommen) aus Teilen der tropischen und subtropischen Regionen auf, in denen Malaria verbreitet ist oder war. Ein Viertel aller Menschen afrikanischer Herkunft südlich der Sahara sind Träger des Gens.
Wir alle erben zwei Kopien (Allele) des Hämoglobin-beta-Gens. Jeder Elternteil erhält eine. Manche Menschen sind heterozygot: Sie haben die Sichelmutation in einer Kopie und die andere Kopie ist normal. Solche Menschen werden Sichelmerkmal oder Träger genannt. Menschen mit Sichel-Eigenschaft sind resistenter gegen Malaria als normale Menschen.
Wenn beide Allele eines Gens ähnlich (homozygot) sind, hat eine Person mutierte Kopien oder beide normal. Wenn beide Allele die Mutation aufweisen, verursacht sie die vollständige Erkrankung. In Malaria-gefährdeten Gebieten sterben normale Menschen häufig an Malaria, oft bevor sie Kinder bekommen haben. Diejenigen mit beiden Kopien mit Sichelmutation sterben an Sichelkrankheit, bevor sie sich fortpflanzen können. Aber die Heterozygoten haben eine bessere Überlebenschance (und haben mehr Kinder) als beide homozygoten Gruppen. Daher ist die Vererbung der Krankheit ein Beispiel für einen "heterozygoten Vorteil".
Bei der vollen (homozygoten) Krankheit verkürzt sich die Lebenserwartung, in vormodernen Gesellschaften ist sie sogar fast tödlich. Sir Cyril Clarke sagte (mit Bezug auf Ostafrika in den 1960er Jahren): "Fast alle Kinder (mit Sichelzellanämie) werden im Säuglingsalter sterben". p25. Studien in den modernen U.S.A. berichten von einer durchschnittlichen Lebenserwartung von 42 Jahren für Männer und 48 Jahren für Frauen.



Verwandte Artikel
Autor
AlegsaOnline.com Sichelzellenanämie Leandro Alegsa
URL: https://de.alegsaonline.com/art/90200
Quellen
- nejm.org : nejm.org/doi/full/10.1056/NEJM200006223422502
- jci.org : "The impact of malaria parasitism: from corpuscles to communities"
- doi.org : 10.1172/JCI38307
- ncbi.nlm.nih.gov : 2735907
- pubmed.ncbi.nlm.nih.gov : 19729847
- content.nejm.org : "Mortality in sickle cell disease. Life expectancy and risk factors for early death"
- doi.org : 10.1056/NEJM199406093302303
- worldcat.org : 0028-4793
- pubmed.ncbi.nlm.nih.gov : 7993409