Cycloadditionen: Pericyclische Reaktionen erklärt (Diels-Alder, 1,3-Dipolar)
Cycloadditionen verständlich erklärt: Mechanismen, Diels–Alder und 1,3‑dipolare Reaktionen, Beispiele, Regelkunde und Reaktionsmechanik für Studium & Forschung.
Eine Cycloaddition ist eine chemische Reaktion zwischen Reaktanten mit Doppelbindungen, die durch eine Ringstruktur ersetzt werden. Es handelt sich um eine pericyclische chemische Reaktion, bei der sich "zwei oder mehr ungesättigte Moleküle (oder Teile desselben Moleküls) mit der Bildung eines cyclischen Addukts verbinden, in dem es zu einer Nettoreduktion der Bindungsvielfalt kommt". Es handelt sich um eine zyklische Reaktion: Es entsteht ein neuer Ring aus Atomen. Pericyclische Reaktionen laufen in der Regel konzertiert ab, das heißt, Bindungen werden in einem einzigen, zusammenhängenden Schritt umorganisiert; dies führt oft zu hoher Stereospezifität der Produkte. Die Betrachtung der beteiligten π-Orbitale und ihrer Symmetrien (Woodward–Hoffmann-Regeln) erklärt, welche Cycloadditionen thermisch oder photochemisch erlaubt sind.
Cycloadditionen werden nach der Grundgröße der Moleküle benannt, die zusammengebracht werden. Damit wäre die Diels-Alder-Reaktion eine [4 + 2]-Cycloaddition und die 1,3-dipolare Cycloaddition eine [3 + 2]-Cycloaddition. Diese Art von Reaktion ist eine unpolare Additionsreaktion.
Bildergalerie
6 Bilder





Grundprinzipien und Mechanismus
Bei pericyclischen Cycloadditionen erfolgt der Elektronentransfer in einem zyklischen Übergangszustand. Wichtige Konzepte sind:
- Frontier-MO-Theorie (FMO): Die Wechselwirkung zwischen dem HOMO eines Reaktanten und dem LUMO des anderen kontrolliert Reaktivität, Regio- und Stereoselektivität. Ein elektronreiches Diene und ein elektronarmes Dienophil beschleunigen beispielsweise die Diels-Alder-Reaktion.
- Woodward–Hoffmann-Regeln: Bestimmen, welche Kombinationen von suprafazialen/antarafazialen Verschiebungen thermisch bzw. photochemisch erlaubt sind. Klassische [4+2]-Diels–Alder-Reaktionen sind thermisch allowed (suprafazial an beiden Komponenten).
- Konzertiert vs. schrittweise: Viele Cycloadditionen sind konzertiert; unter bestimmten Bedingungen (z. B. bei stark substituierten Systemen oder besonderen Substituenten) können zwischengeschaltete Radikal- oder Ionenzustände auftreten.
Stereochemie und Regioselektivität
Cycloadditionen sind häufig stereospezifisch: stereochemische Informationen in den Edukten werden in das Produkt überführt. Wichtige Aspekte sind:
- Endo-/Exo-Regel (bei Diels–Alder): Bei vielen dienophile Substituenten wird das endo-Produkt bevorzugt, weil sekundäre Wechselwirkungen im Übergangszustand stabilisierend wirken (klassisch: Cyclopentadien + Maleinsäureanhydrid → überwiegend endo-Produkt).
- Regioselektivität: Elektronenziehende bzw. -schiebende Substituenten führen zu bevorzugter Orientierung, die sich mit Hilfe der FMO-Analyse vorhersagen lässt.
- Stereospezifität: Trans- bzw. cis-Anordnungen in den Ausgangsmaterialien führen meist zu entsprechend trans-/cis-substituierten Produkten.
Typen von Cycloadditionen — Beispiele
- Diels–Alder (4+2): Diene + Dienophil → cyclohexenartige Produkte. Häufig eingesetzt in der Synthese komplexer Naturstoffe. Typisches Beispiel: Cyclopentadien + Maleinsäureanhydrid.
- 1,3-Dipolare Cycloaddition (3+2): Ein 1,3-Dipol (z. B. Nitrilon, Nitriloxid, Azomethinylid) reagiert mit einem Dipolarophil (Alken/Alkin) zu fünfgliedrigen Heterocyclen. Beispiele: Bildung von Isoxazolen, Isoxazolidinen oder 1,2,3-Triazolen.
- Cheletrope Reaktionen: Sonderfall, bei dem beide neuen Bindungen zu demselben Atom gebildet werden (z. B. SO2-Addition an ein Cyclopentadienylid).
- Azid-Alkin-Cycloaddition (Huisgen, „Click“): Die thermische 1,3-dipolare Cycloaddition von Aziden und Alkinen liefert 1,2,3-Triazole; die kupferkatalysierte Variante (CuAAC) ist eine extrem nützliche, schnelle und regioselektive Methode in der „Click“-Chemie (führt bevorzugt zu 1,4-substituierten Triazolen).
Katalyse, Bedingungen und praktische Hinweise
- Lewis-Säuren (z. B. AlCl3, BF3·OEt2) aktivieren Dienophile, indem sie deren LUMO absenken und so Reaktionsgeschwindigkeit und Selektivität erhöhen.
- Temperatur und Lösungsmittel: Höhere Temperaturen beschleunigen oft die Reaktion, können aber Selektivität verringern; polare Lösungsmittel können polare Charaktere verstärken und so Reaktionsraten beeinflussen.
- Photochemische Aktivierung: Unter Bestrahlung können sonst thermisch verbotene Pfade zugänglich werden oder umgekehrt die Reaktivität verändern (andere Orbital-Symmetriebedingungen gelten).
- Katalysierte Varianten: Neben Lewis-Säuren gibt es Metallkatalysen und organokatalytische Konzepte, die z. B. regio- und enantioselektive Cycloadditionen ermöglichen.
Anwendungen
Cycloadditionen sind in der organischen Synthese außerordentlich wichtig:
- Synthese komplexer Naturstoffe und Pharmaka durch Aufbau ringförmiger Gerüste in wenigen Schritten.
- Materialwissenschaften: Herstellung von funktionellen Polymeren und Vernetzungen (z. B. durch Diels–Alder-Click-Verknüpfungen).
- Biokonjugation: CuAAC als robustes Werkzeug zum Kopplen von Biomolekülen.
Kurze Zusammenfassung
- Cycloadditionen bauen Ringe auf und laufen oft konzertiert (pericyclisch) ab.
- Die Bezeichnung [m+n] gibt die Anzahl der π-Elektronen bzw. Atome der addierenden Fragmente an (z. B. [4+2], [3+2]).
- FMO-Theorie und Woodward–Hoffmann-Regeln erklären Reaktivität, Selektivität und erlaubte Reaktionspfade.
- Praktisch wichtig sind Diels–Alder, 1,3-dipolare Cycloaddition und die kupferkatalysierte Azid‑Alkin‑Cycloaddition („Click“).
Bei speziellen Fragestellungen (z. B. detaillierte FMO-Analysen, stereochemische Vorhersagen für konkrete Substrate oder experimentelle Bedingungen) kann ich gern konkrete Beispiele und Mechanismusdiagramme erläutern.
Reaktionsmechanismus
Hitze kann dazu führen, dass die Doppelbindungen einen Ring bilden. Thermische Cycloadditionen haben normalerweise (4n + 2) π Elektronen, die am Ausgangsmaterial beteiligt sind, für eine ganze Zahl n. Wegen der Orbitalsymmetrie sind die meisten Cycloadditionen suprafazial-suprafazial. Selten sind sie antarafazial-antarafazial. Es gibt einige Beispiele für thermische Cycloadditionen, die 4n π Elektronen haben (zum Beispiel die [2 + 2] Cycloaddition). Diese verlaufen in einem suprafazial-antarafazialen Sinn. Zum Beispiel hat die Dimerisierung von Keten eine orthogonale Menge von p-Orbitalen. Diese p-Orbitale ermöglichen es, dass die Reaktion mit einem gekreuzten Übergangszustand abläuft.
Licht kann auch dazu führen, dass die Doppelbindungen einen Ring bilden. Cycloadditionen, an denen 4n π Elektronen beteiligt sind, können auch als Folge einer photochemischen Aktivierung auftreten. Hier führt eine Komponente eine Elektronenbewegung vom höchsten besetzten Molekülorbital (HOMO) (π-Bindung) zum niedrigsten unbesetzten Molekülorbital (LUMO) (π*-Antibindung) durch. Nachdem das Elektron in das höhere Orbital befördert wurde, ermöglicht die Orbitalsymmetrie, dass die Reaktion suprafazial-suprafazial abläuft. Ein Beispiel ist die DeMayo-Reaktion. Ein weiteres Beispiel ist unten dargestellt, die photochemische Dimerisierung von Zimtsäure.
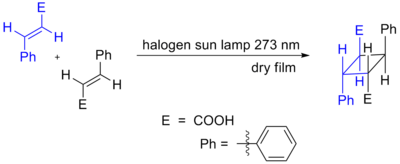
Beachten Sie, dass nicht alle photochemischen (2+2) Zyklisierungen Zyklloadditionen sind; von einigen ist bekannt, dass sie über radikale Mechanismen ablaufen.
Einige Cycloadditionen anstelle von π Anleihen funktionieren über gespannte Cyclopropanringe; da diese einen signifikanten π Charakter haben. Ein Analogon für die Diels-Alder-Reaktion ist zum Beispiel die Quadricyclan-DMAD-Reaktion:
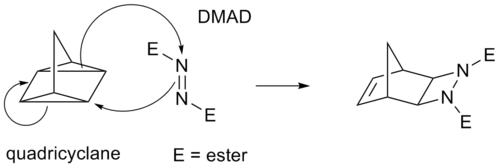
In der (i+j+...) Cycloaddition-Notation beziehen sich i und j auf die Anzahl der an der Cycloaddition beteiligten Atome. In dieser Notation ist eine Diels-Alder-Reaktion eine (4+2)-Cycloaddition und eine 1,3-dipolare Addition wie der erste Schritt in der Ozonolyse ist eine (3+2)-Cycloaddition. Diese Notation verwendet Klammern. Die von der IUPAC bevorzugte Schreibweise mit [i+j+...] zählt jedoch Elektronen und keine Atome. Sie verwendet eckige Klammern. In dieser Schreibweise werden sowohl die Diels-Alder-Reaktion als auch die dipolare Reaktion zu einer [4+2]-Cycloaddition. Die Reaktion zwischen Norbornadien und einem aktivierten Alkin ist eine [2+2+2]-Cycloaddition.
Arten von Cycloadditionen
Diels-Alder-Reaktionen
Die Diels-Alder-Reaktion ist eine [4+2]-Cycloadditions-Reaktion.
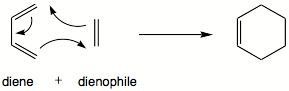
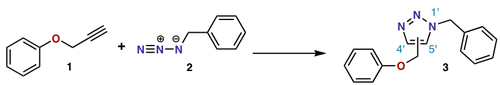
Huisgen-Zykloads
Die Huisgen-Cycloadditions-Reaktion ist eine [2+3]-Cycloaddition.
Nitron-Olefin-Cycload-Zustand
Die Nitron-Olefin-Cycloaddition ist eine [3+2]-Cycloaddition.
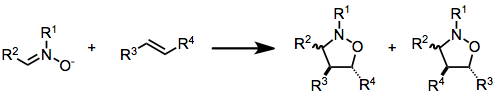
Formale Cycload-Bedingungen
Cycloadditionen haben oft metallkatalysierte und stufenweise radikale Analoga, jedoch handelt es sich dabei streng genommen nicht um pericyclische Reaktionen. Wenn geladene oder radikalische Zwischenprodukte an einer Cycloaddition beteiligt sind, oder wenn das Ergebnis der Cycloaddition in einer Reihe von Reaktionsschritten gefunden wird, werden sie manchmal als formale Cycloadditionen bezeichnet, um einen Unterschied zu echten pericyclischen Cycloadditionen zu machen.
Ein Beispiel für eine formale [3+3]-Cycloaddition zwischen einem zyklischen Enon und einem Enamin, die durch n-Butyllithium katalysiert wird, ist eine Storchen-Enamin / 1,2-Additions-Kaskadenreaktion:
![Intermolecular Formal [3+3] Cycloaddition Reaction](https://www.alegsaonline.com/image/600px-3%2B3-cycloaddition.svg.png)
Fragen und Antworten
F: Was ist eine Cycloaddition?
A: Eine Cycloaddition ist eine chemische Reaktion zwischen Reaktionspartnern mit Doppelbindungen, die durch eine Ringstruktur ersetzt werden.
F: Welche Art von chemischer Reaktion ist eine Cycloaddition?
A: Eine Cycloaddition ist eine perizyklische chemische Reaktion, bei der "zwei oder mehr ungesättigte Moleküle (oder Teile desselben Moleküls) sich unter Bildung eines zyklischen Addukts verbinden, bei dem es zu einer Nettoverringerung der Bindungsvielfalt kommt".
F: Was geschieht bei einer Cycloadditionreaktion?
A: Eine Cycloaddition ist eine Zyklisierungsreaktion: Sie bildet einen neuen Ring aus Atomen.
F: Wie werden Cycloadditionen benannt?
A: Cycloadditionen werden nach der Grundgröße der Moleküle benannt, die zusammengebracht werden.
F: Was ist die Diels-Alder-Reaktion?
A: Die Diels-Alder-Reaktion ist eine [4 + 2]-Cycloaddition.
F: Was ist die 1,3-dipolare Cycloaddition?
A: Die 1,3-dipolare Cycloaddition ist eine [3 + 2]-Cycloaddition.
F: Welche Art von Reaktion ist eine Cycloaddition?
A: Eine Cycloaddition ist eine unpolare Additionsreaktion.
Verwandte Artikel
Autor
AlegsaOnline.com Cycloadditionen: Pericyclische Reaktionen erklärt (Diels-Alder, 1,3-Dipolar) Leandro Alegsa
URL: https://de.alegsaonline.com/art/24866
Quellen
- goldbook.iupac.org : Cycloaddition
- doi.org : 10.1021/ed083p940
- doi.org : 10.1002/anie.200603302
- pubmed.ncbi.nlm.nih.gov : 17146819