Woodward-Fieser-Regeln: UV-Absorptionsmaxima (λmax) in der organischen Chemie
Woodward‑Fieser‑Regeln: Verständliche Vorhersage von UV‑Absorptionsmaxima (λmax) für Chromophore, konjugierte Diene und Carbonylverbindungen in der organischen Chemie.
Woodwards Regeln sind eine Reihe empirischer Richtlinien dafür, wie organisch-chemische Verbindungen ultraviolettes Licht absorbieren. Sie geben insbesondere Aufschluss über die Wellenlänge des Absorptionsmaximums (Symbol λmax) in einem ultraviolett-sichtbaren (UV) Spektrum einer Verbindung. Die Regeln sind nach Robert Burns Woodward benannt. Er war Professor an der Harvard University und erhielt 1965 den Nobelpreis für Chemie. Weil Louis Fieser wichtige Ergänzungen und Tabellen lieferte, werden die Regeln oft als Woodward-Fieser-Regeln bezeichnet.
Grundprinzipien
Die Woodward-Fieser-Regeln basieren auf dem Grundsatz, dass die Position des Absorptionsmaximums durch drei Hauptfaktoren bestimmt wird:
- der Typ und die Länge des Chromophors (z. B. konjugierte Carbonylgruppe, konjugiertes Dien, Polyen),
- Art und Anzahl der Substituenten bzw. Auxochrome am Chromophor (Elektronendonoren wie -OH, -OR, Aminogruppen; alkylische Reste; exocyclische Doppelbindungen u. a.),
- Änderungen durch das Lösungsmittel (solvatochrome Effekte) und andere Umgebungsfaktoren.
Als Ergebnis liefern die Regeln für einen gegebenen Chromophortyp einen Basiswert für λmax und geben dann tabellarisch hinzuzufügende Beiträge (in nm) für verschiedene Substituenten und strukturelle Besonderheiten an. Man erhält so eine schnelle Näherung für das erwartete Absorptionsmaximum.
Typische Chromophore und Anwendungsbeispiele
Besonders häufig angewendet werden die Regeln für:
- konjugierte Carbonylverbindungen (α,β-ungesättigte Ketone, Aldehyde, Ester usw.): Hier bestimmt die Carbonylgruppe in Kombination mit der konjugierten Doppelbindung das Chromophor. Substituenten und zusätzliche Konjugation verschieben λmax meist bathochrom (zu längeren Wellenlängen).
- konjugierte Diene: offenkettige und ringförmige Diene (homoannulare vs. heteroannulare) zeigen charakteristische Basiswerte und additive Effekte durch Alkylgruppen oder exocyclische Doppelbindungen.
- Polyen: Mit zunehmender Anzahl konjugierter Doppelbindungen verschiebt sich λmax deutlich in den langwelligeren Bereich; die Regeln liefern grobe Abschätzungen für diese Verschiebung.
Wie man λmax abschätzt (Schritt-für-Schritt)
- Chromophor identifizieren (z. B. α,β-ungesättigtes Ketonsystem, 1,3-Dien usw.).
- Den für diesen Chromophortyp angegebenen Basiswert für λmax aus einer Woodward‑Fieser-Tabelle heranziehen.
- Für jeden relevanten Substituenten, jede zusätzliche Konjugationseinheit oder spezielle Strukturmerkmale die in der Tabelle angegebenen Zusatzwerte aufsummieren.
- Einen Korrekturfaktor für das verwendete Lösungsmittel oder für besondere Wechselwirkungen (z. B. intramolekulare Wasserstoffbrücken, starken mesomeren Effekt) berücksichtigen.
- Die Summe ergibt eine Näherung für das erwartete λmax im UV‑/Vis‑Spektrum.
Wichtige Einflüsse und Einschränkungen
- Empirischer Charakter: Die Regeln sind Näherungen, die auf experimentellen Tabellen beruhen. Sie funktionieren gut für einfache, klar definierte Chromophore, sind aber weniger zuverlässig bei stark substituierten, sterisch gehinderten oder sehr polarisierten Systemen.
- Auxochrome: Gruppen, die die Elektronendichte verändern (z. B. -OH, -OR, -NR2), führen meist zu einem bathochromen Shift; Alkylgruppen wirken schwächer über hyperkonjugative Effekte.
- Konjugationsverlängerung: Jede zusätzliche konjugierte Doppelbindung verschiebt das Absorptionsmaximum nach längeren Wellenlängen (bathochrom), oft stärker als einzelne Alkylsubstituenten.
- Solvent- und Temperaturabhängigkeit: Polarere Lösungsmittel können durch Stabilisierung von Grund- oder angeregten Zuständen zu Verschiebungen führen (solvatochrome Effekte).
- Spezialfälle: Heteroatome, aromatische Systeme, intramolekulare Wechselwirkungen oder starke intramolekulare Konjugation können die Vorhersagegenauigkeit reduzieren; in solchen Fällen sind ergänzende Daten oder Berechnungen empfehlenswert.
Moderne Alternativen und praktische Hinweise
Die Woodward‑Fieser‑Regeln sind nach wie vor nützlich für schnelle Abschätzungen und zur Interpretation von UV‑Spektren in Lehr- und Laborsituationen. Für präzisere Vorhersagen oder komplexe Moleküle stehen heute jedoch quantenchemische Methoden (z. B. TD‑DFT) zur Verfügung, die UV/Vis‑Spektren auf Basis elektronischer Struktur berechnen. In der Praxis empfiehlt es sich, die Woodward‑Fieser‑Schätzung als ersten Anhaltspunkt zu verwenden und bei Bedarf experimentelle Messungen oder computergestützte Rechnungen hinzuzuziehen.
Zusammenfassend: Die Woodward‑Fieser‑Regeln verbinden einfache, empirische Basiswerte für typische Chromophore mit additiven Beiträgen durch Substituenten und Umgebungsfaktoren und bieten so eine schnelle Methode zur Abschätzung des λmax in UV/Vis‑Spektren organischer Verbindungen.
Umsetzung
Ein Satz von Woodward-Fieser-Regeln für Diene ist in Tabelle eins dargestellt. Ein Dien ist entweder homo-ringförmig, wobei beide Doppelbindungen in einem Ring enthalten sind, oder hetero-ringförmig, wobei zwei Doppelbindungen zwischen zwei Ringen verteilt sind.
| Basiswert für heteroannulares Dien | 214 |
| Basiswert für homoannulares Dien | 253 |
| Inkremente | |
| Doppelbindungserweiternde Konjugation | + 30 |
| Alkyl-Substituent oder Ring-Rest | + 5 |
| Exozyklische Doppelbindung | + 5 |
| Acetat-Gruppe | + 0 |
| Äther-Gruppe | + 6 |
| Thioether-Gruppe | + 30 |
| Brom, Chlor | + 5 |
| sekundäre Amin-Gruppe | + 60 |
| Tabelle 1. Regeln für die Wellenlänge der maximalen | |
Diese Regeln sagen das UV-Absorptionsmaximum von Verbindungen voraus. Hier sind zwei Beispiele:
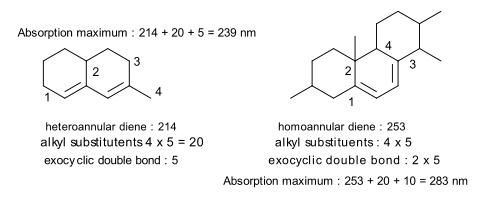
In der Verbindung auf der linken Seite beträgt der Basiswert 214 nm (ein heteroannulares Dien). Diese Diengruppe hat 4 Alkylsubstituenten (markiert mit 1,2,3,4) und die Doppelbindung in einem Ring ist exocyclisch zu dem anderen (5 nm für eine exocyclische Doppelbindung hinzugefügt). In der Verbindung auf der rechten Seite ist das Dien homoannulär mit 4 Alkylsubstituenten. Beide Doppelbindungen im zentralen B-Ring sind in Bezug auf die Ringe A und C exocyclisch.
Fragen und Antworten
F: Was sind die Woodwardschen Regeln?
A: Die Woodward'schen Regeln sind eine Reihe von Regeln darüber, wie organische chemische Verbindungen ultraviolettes Licht absorbieren.
F: Worüber geben die Woodward'schen Regeln Auskunft?
A: Die Woodwardschen Regeln geben Auskunft über die Wellenlänge des Absorptionsmaximums (Symbol λmax) in einem ultraviolett-sichtbaren (UV) Spektrum einer Verbindung.
F: Nach wem sind die Regeln benannt?
A: Die Regeln sind nach Robert Burns Woodward benannt.
F: Was war der Beruf von Robert Burns Woodward?
A: Robert Burns Woodward war Professor an der Harvard University.
F: Wofür hat Robert Burns Woodward den Nobelpreis erhalten?
A: Robert Burns Woodward erhielt 1965 den Nobelpreis für Chemie.
F: Wer wird noch mit den Woodward-Fieser-Regeln geehrt?
A: Die Woodward-Fieser-Regeln ehren auch Louis Fieser.
F: Worauf beruhen die Woodward-Regeln bei Vorhersagen?
A: Die Woodward-Regeln basieren auf der Art der vorhandenen Chromophore, den Substituenten an den Chromophoren und den Veränderungen durch das Lösungsmittel.
Verwandte Artikel
Autor
AlegsaOnline.com Woodward-Fieser-Regeln: UV-Absorptionsmaxima (λmax) in der organischen Chemie Leandro Alegsa
URL: https://de.alegsaonline.com/art/108992
Quellen
- doi.org : 10.1021/ja01849a066
- doi.org : 10.1021/jo01164a003
- pubmed.ncbi.nlm.nih.gov : 18106021
- chemistry.ccsu.edu : "Woodward's Rules for Conjugated Carbonyl Compounds"
- cem.msu.edu : "UV-Visible Spectroscopy"
- chemistry.ccsu.edu : "Woodward-Fieser Rules for Dienes"