Pulmonale Hypertonie (Lungenhochdruck): Ursachen, Symptome & Therapie
Pulmonale Hypertonie (Lungenhochdruck): Ursachen, Symptome & Therapie. Erkennen, behandeln und lebensrettende Optionen von Medikamenten bis Transplantation verständlich erklärt.
Pulmonale Hypertonie oder PH ist eine Erkrankung, bei der ein hoher Blutdruck in der Lunge besteht. Dieser erhöhte Druck betrifft die Lungenarterien und macht es schwieriger, Luft aufzunehmen und zu verarbeiten, sodass das Atmen anstrengender wird. Manche Betroffene benötigen zusätzlichen Sauerstoff, weil der Gasaustausch eingeschränkt ist. Die Erkrankung kann Schwindel, anhaltende Müdigkeit und in einigen Fällen Ohnmachtsanfälle verursachen; einige Menschen werden leicht ohnmächtig, vor allem bei Belastung. Weil das Herzen gegen einen höheren Widerstand in der Lunge pumpen muss, kann es überlastet werden und im Verlauf krankhafte Veränderungen entwickeln. In schweren, fortgeschrittenen Fällen kann eine Lungentransplantation oder sogar eine Herz‑Lungen‑Transplantation notwendig sein. Pulmonale Hypertonie ist oft auch unter dem vollständigen Begriff pulmonal‑arterielle Hypertonie bekannt; in der Praxis werden die Abkürzungen PAH, PH oder PHA verwendet.
Bildergalerie
9 Bilder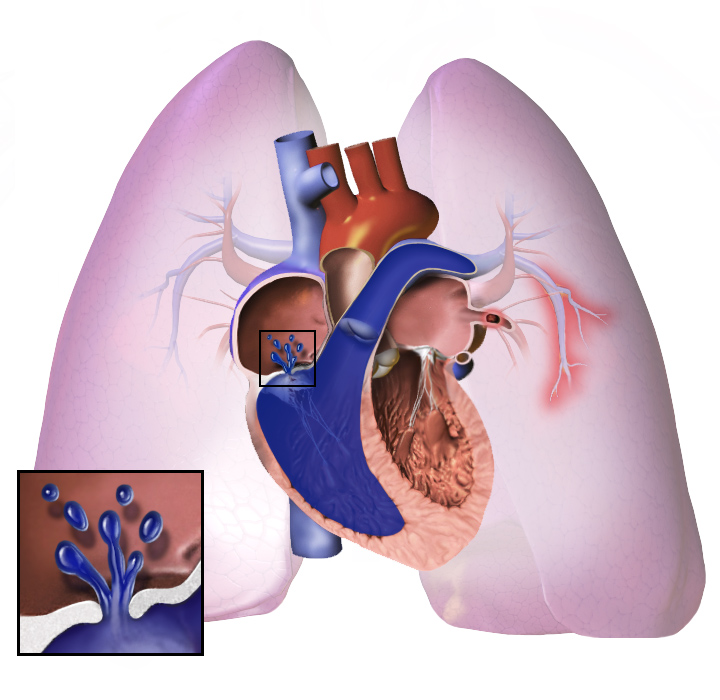






Ursachen
Pulmonale Hypertonie kann unterschiedliche Ursachen haben. Wichtige Gruppen sind:
- Erkrankungen des linken Herzens (z. B. Herzinsuffizienz, Klappenfehler), die den Druck in den Lungengefäßen erhöhen.
- Chronische Lungenerkrankungen (z. B. COPD, interstitielle Lungenerkrankungen), die den Sauerstofftransport verschlechtern.
- Chronische Blutgerinnsel in den Lungenarterien (chronisch thromboembolische pulmonale Hypertonie, CTEPH).
- Erkrankungen des Bindegewebes (z. B. Sklerodermie), Medikamente oder Drogen, Infektionen (z. B. HIV), Lebererkrankungen (portale Hypertonie) und angeborene Herzfehler.
- In vielen Fällen bleibt die Ursache zunächst unklar (idiopathische PAH).
Symptome
Die Beschwerden beginnen oft schleichend und verschlechtern sich mit der Zeit. Häufige Symptome sind:
- Belastungsbedingte Atemnot (Dyspnoe)
- Frühe Erschöpfung und reduzierte Leistungsfähigkeit
- Ohnmachtsanfälle oder Schwindel, besonders bei Anstrengung
- Brustschmerzen oder Druckgefühl
- Bein‑ oder Bauchschwellungen (Ödeme) bei zunehmender Herzbelastung
- Herzklopfen oder Rhythmusstörungen
Diagnose
Eine frühzeitige Abklärung ist wichtig. Häufige Schritte zur Diagnosesicherung:
- Körperliche Untersuchung und Anamnese (Symptome, Vorerkrankungen, Medikamente).
- Ruhe‑EKG und Echokardiographie (Ultraschall des Herzens) als erste nicht‑invasive Untersuchungen.
- Die Rechtsherzkatheteruntersuchung ist der Goldstandard zur Messung des Drucks in den Lungenarterien und zur endgültigen Bestätigung.
- Lungenfunktionsprüfung, Blutgasanalyse, Bildgebung wie CT der Lunge und Perfusionsszintigrafie (V/Q‑Scan) – besonders wichtig zum Ausschluss einer CTEPH.
- Laboruntersuchungen, BNP/NT‑proBNP zur Abschätzung der Herzbelastung sowie gezielte Tests je nach Verdacht (z. B. Autoantikörper, HIV‑Test, Leberuntersuchungen).
Behandlung
Die Therapie richtet sich nach der Ursache und dem Schweregrad. Ziele sind Symptomlinderung, Verbesserung der Belastbarkeit und Verzögerung des Krankheitsverlaufs.
- Allgemeine Maßnahmen: Sauerstoffgabe bei Bedarf, körperliche Reha/Training unter fachlicher Anleitung, Salzreduktion und Gewichtsmanagement, Impfungen (z. B. gegen Grippe und Pneumokokken).
- Medikamentöse Basistherapie: Diuretika zur Reduktion von Ödemen, Antikoagulation bei bestimmten Ursachen (z. B. CTEPH), und Behandlung zugrundeliegender Erkrankungen (z. B. Herzkrankheiten, Lebererkrankung).
- Spezifische PAH‑Therapien: Endothelinrezeptorantagonisten, Phosphodiesterase‑5‑Hemmer, Prostazyklin‑Analoga oder Prostacyclinrezeptor‑Agonisten sowie Guanylatcyclase‑Stimulatoren. Die Auswahl und Kombination erfolgt in spezialisierten Zentren.
- Bei CTEPH sind spezielle Verfahren möglich: chirurgische Pulmonalendarteriektomie (Thromboendarteriektomie) oder interventionelle Ballon‑Pulmonalarterienangioplastie.
- Fortgeschrittene Stadien: Bei mangelndem Ansprechen auf Therapie kann eine Lungentransplantation oder Herz‑Lungen‑Transplantation erforderlich sein.
Verlauf und Prognose
Die Prognose hängt stark von der zugrundeliegenden Ursache, dem Zeitpunkt der Diagnose und dem Ansprechen auf die Therapie ab. Frühe Diagnose und Behandlung in einem spezialisierten Zentrum verbessern die Chancen deutlich. Regelmäßige Kontrollen (Klinik, Echokardiographie, Belastungstests, Labor) sind wichtig, um Therapieerfolge zu überwachen und Anpassungen vorzunehmen.
Alltagsmaßnahmen und Lebensstil
Betroffene profitieren von gezielter körperlicher Aktivität nach individueller Absprache, guter Therapieadhärenz, Vermeidung von Situationen mit niedrigem Sauerstoffangebot (z. B. sehr große Höhen) und enger Abstimmung bei Reisen. Schwangerschaft ist bei vielen Formen der PH mit erheblichen Risiken verbunden und sollte im Vorfeld mit Fachärzten besprochen werden.
Wann dringend Ärztliche Hilfe suchen
Suchen Sie sofort ärztliche Hilfe oder den Notruf, wenn plötzliche oder stark zunehmende Atemnot, Ohnmachtsanfälle, neu auftretende starke Brustschmerzen oder plötzlich zunehmende Schwellungen auftreten. Bei bekannter pulmonaler Hypertonie ist die Betreuung durch ein spezialisiertes Zentrum ratsam.
Wenn Sie den Verdacht auf pulmonale Hypertonie haben oder Symptome zunehmen, sprechen Sie mit Ihrem Hausarzt oder Kardiologen/Pneumologen. Eine gezielte Abklärung kann lebenswichtig sein und Therapieoptionen eröffnen.
Anzeichen und Symptome
Menschen mit pulmonaler Hypertonie haben Atembeschwerden. Außerdem ermüden sie leicht. Einige von ihnen fallen auch leicht in Ohnmacht. Sie können Brustschmerzen haben. Einige Patienten haben Schwellungen an den Füßen und Knöcheln. Diese Symptome verschlimmern sich bei Bewegung oder harter Arbeit.
Da viele Krankheiten das Atmen erschweren können, muss ein Arzt den Hintergrund des Patienten kennen lernen. Dies hilft dem Arzt, den Patienten zu behandeln, auch wenn der Patient eine andere Krankheit hat. Der Arzt führt auch mehrere Tests durch. Bei pulmonaler Hypertonie klingt das Herz anders. Ein Test ist die Messung des Blutdrucks in der Lungenarterie, dem Blutgefäß, das vom Herzen zu den Lungen führt.
Um die Ursache festzustellen, wird der Arzt in der Regel eine gründliche Anamnese durchführen. Eine detaillierte Familienanamnese wird erstellt, um festzustellen, ob die Krankheit familiär bedingt sein könnte. Eine Anamnese der Exposition gegenüber Kokain, Methamphetamin, Alkohol, die zu einer Leberzirrhose führt, und Rauchen, das zu einem Emphysem führt, wird als signifikant angesehen. Es wird eine körperliche Untersuchung durchgeführt, um nach typischen Anzeichen einer pulmonalen Hypertonie zu suchen, darunter ein lautes P2 (pulmonales Klappenschlussgeräusch), (para)sternale Hebung, juguläre Venenauftreibung, Pedalödem, Aszites, hepatojugulärer Reflux, Keulenbildung usw.
Was mit dem Körper schief geht
Bei der pulmonalen Hypertonie werden die Blutgefässe in der Lunge zu eng. Der Blutdruck in der Lunge wird hoch. Das Herz arbeitet sehr hart, um Blut durch die engen Blutgefässe zu pumpen. Später werden die Blutgefässe in der Lunge hart und dick. Das Herz muss härter arbeiten.
Das Herz kann so hart arbeiten, dass es krank wird. Dies wird als Herzinsuffizienz bezeichnet. Das kranke Herz kann das Blut nicht gut pumpen. Es gelangt weniger Blut in die Lungen, so dass das Blut weniger Sauerstoff erhält. Dadurch wird das Atmen erschwert. Dies verschlimmert sich bei körperlicher Anstrengung oder harter Arbeit.
Ursachen
Die häufigste Ursache der pulmonalen Hypertonie ist die Linksherzinsuffizienz. Diese verursacht eine pulmonal-venöse Hypertonie. Dies führt zu einem Lungenödem oder einer Flüssigkeitsansammlung in der Lunge.
Viele Erkrankungen können eine pulmonal-arterielle Hypertonie (PAH) verursachen.
- Lungenkrankheiten, bei denen das Blut weniger Sauerstoff hat, wie zum Beispiel
· chronisch obstruktive Lungenerkrankung oder COPD
· interstitielle Lungenerkrankung
· Pickwickian-Syndrom
- Probleme des Immunsystems, wie zum Beispiel
· AIDS
· Sklerodermie
· andere Autoimmunerkrankungen
- Leberprobleme
· Zirrhose
· Portal-Hypertonie
- andere Ursachen
· Schlafapnoe
· Einnahme von Tabletten zum Abnehmen, wie Fen-Phen, Aminorex, Fenfluramin (Pondimin) und Phentermin
· angeborene Herzkrankheit
· Schilddrüsenerkrankungen,
· Drogen wie Kokain nehmen
· möglicherweise Humane Herpesviren 8
Wenn eine Person an pulmonaler Hypertonie ohne andere Ursache leidet, wird dies als idiopathische pulmonal-arterielle Hypertonie oder IPAH bezeichnet.
Wenn eine Familienanamnese vorliegt, wird die Erkrankung als familiäre pulmonal-arterielle Hypertonie (FPAH) bezeichnet. IPAH und FPAH werden heute als genetische Störungen betrachtet, die mit Mutationen im BMPR2-Gen, das einen Rezeptor für knochenmorphogenetische Proteine kodiert, sowie im 5-HT(2B)-Gen, das einen Serotoninrezeptor kodiert, in Verbindung stehen.
In der Medizin versteht man unter pulmonaler Hypertonie (PH) einen Anstieg des Blutdrucks in der Lungenarterie oder den Lungengefässen, der zu Kurzatmigkeit, Schwindel, Ohnmacht und anderen Symptomen führt, die durch die Anstrengung verschlimmert werden. Je nach Ursache kann es sich bei der pulmonalen Hypertonie um eine schwere Erkrankung mit deutlich verminderter Belastungstoleranz und Rechtsherzinsuffizienz handeln. Sie wurde erstmals 1891 von Dr. Ernst von Romberg identifiziert. Sie kann eine von fünf verschiedenen Arten sein: arterielle, venöse, hypoxische, thromboembolische oder sonstige.
Obwohl die Begriffe primäre pulmonale Hypertonie (Bedeutung von unbekannter Ursache) und sekundäre pulmonale Hypertonie (Bedeutung aufgrund einer anderen Erkrankung) in Materialien, die an Patienten und die breite Öffentlichkeit verteilt werden, immer noch vorkommen, sind diese Begriffe in der medizinischen Literatur weitgehend aufgegeben worden. Diese Veränderung ist darauf zurückzuführen, dass die ältere dichotome Klassifikation weder die Pathophysiologie noch das Ergebnis widerspiegelte. Sie führte zu fehlerhaften Therapieentscheidungen, d.h. nur "primäre" pulmonale Hypertonie zu behandeln. Dies wiederum führte bei vielen Patienten, die als "sekundäre" pulmonale Hypertonie bezeichnet wurden, zu therapeutischem Nihilismus und könnte zu ihrem Tod beigetragen haben. Der Begriff "primäre pulmonale Hypertonie" wurde nun durch "idiopathische pulmonal-arterielle Hypertonie" ersetzt. Die Begriffe "primäre" und "sekundäre" pulmonale Hypertonie sollten nicht mehr verwendet werden. Weitere Einzelheiten finden Sie im Abschnitt "Klassifikation" weiter unten.
Ursachen
Die häufigste Ursache der pulmonalen Hypertonie ist die Linksherzinsuffizienz, die zu einer pulmonal-venösen Hypertonie führt. Dies kann auf eine systolische oder diastolische Fehlfunktion der linken Herzkammer oder auf eine Klappenfunktionsstörung wie Mitralinsuffizienz oder Mitralstenose zurückzuführen sein. Sie manifestiert sich gewöhnlich als Lungenödem.
Zu den häufigen Ursachen der pulmonal-arteriellen Hypertonie (PAH) gehören HIV, Sklerodermie und andere Autoimmunerkrankungen, Zirrhose und portale Hypertonie, Sichelzellanämie, angeborene Herzerkrankungen, Schilddrüsenerkrankungen und andere. Die Einnahme von Gewichtsabnahmepillen wie Fen-Phen, Aminorex, Fenfluramin (Pondimin) und Phentermin führte in der Vergangenheit zur Entwicklung der PAH.
Pathogenese
Unabhängig von der anfänglichen Ursache führt pulmonale Hypertonie zu einer Verengung der Blutgefässe, die mit der Lunge verbunden sind und sich innerhalb der Lunge befinden. Dadurch wird es für das Herz schwieriger, Blut durch die Lungen zu pumpen, so wie es auch schwieriger ist, Wasser durch ein enges statt durch ein breites Rohr fließen zu lassen. Im Laufe der Zeit werden die betroffenen Blutgefässe sowohl steifer als auch dicker, wodurch der Blutdruck in der Lunge weiter ansteigt und der Blutfluss beeinträchtigt wird. Darüber hinaus führt die erhöhte Arbeitsbelastung des Herzens zu einer Verdickung und Vergrößerung der rechten Herzkammer, wodurch das Herz weniger in der Lage ist, Blut durch die Lungen zu pumpen, was zu einer Rechtsherzinsuffizienz führt. Wenn der Blutfluss durch die Lungen abnimmt, erhält die linke Seite des Herzens weniger Blut. Dieses Blut kann auch weniger Sauerstoff transportieren als normal. Daher wird es für die linke Herzhälfte immer schwieriger zu pumpen, um den Rest des Körpers mit ausreichend Sauerstoff zu versorgen, insbesondere bei körperlicher Aktivität.
Diagnose
Da es 5 Haupttypen der pulmonalen Hypertonie geben kann, muss eine Reihe von Tests durchgeführt werden, um die pulmonal-arterielle Hypertonie von der venösen, hypoxischen, thomboembolischen oder verschiedenen Varianten zu unterscheiden.
Es wird eine körperliche Untersuchung durchgeführt, um nach typischen Anzeichen einer pulmonalen Hypertonie zu suchen. Dazu gehören veränderte Herztöne, wie z.B. ein weit gespaltener S2- oder zweiter Herzton, ein lautes P2- oder pulmonales Klappenschlussgeräusch (Teil des zweiten Herztons), (para)sternale Hebung, möglicherweise S3- oder dritter Herzton und pulmonale Regurgitation. Weitere Anzeichen sind jugulär-venöse Dehnung (Erweiterung der Jugularvenen), peripheres Ödem (Schwellung der Knöchel und Füße), Aszites (Bauchschwellung durch Flüssigkeitsansammlung), hepatojugulärer Reflux und Keulenbildung.
Weitere Eingriffe sind erforderlich, um das Vorliegen einer pulmonalen Hypertonie zu bestätigen und andere mögliche Diagnosen auszuschließen. Dazu gehören in der Regel Lungenfunktionstests, Blutuntersuchungen, Elektrokardiographie (EKG), arterielle Blutgasmessungen, Röntgenaufnahmen des Brustkorbs (gefolgt von einem hochauflösenden CT-Scan bei Verdacht auf eine interstitielle Lungenerkrankung) und eine Beatmungs-Perfusion oder V/Q-Scanning zum Ausschluss einer chronisch thromboembolischen pulmonalen Hypertonie. Eine Lungenbiopsie ist in der Regel nicht indiziert, es sei denn, es wird vermutet, dass die pulmonale Hypertonie auf eine zugrunde liegende interstitielle Lungenerkrankung zurückzuführen ist. Lungenbiopsien sind jedoch aufgrund des hohen intrapulmonalen Blutdrucks mit Blutungsrisiken behaftet. Die klinische Besserung wird oft durch einen "Sechs-Minuten-Gehtest" gemessen, d.h. die Strecke, die ein Patient in sechs Minuten zurücklegen kann. Stabilität und Verbesserung dieser Messung korrelieren mit einem besseren Überleben.
Obwohl der pulmonal-arterielle Druck auf der Grundlage der Echokardiographie abgeschätzt werden kann, liefert die Druckprobenentnahme mit einem Swan-Ganz-Katheter die eindeutigste Messung. PAOP und PVR können mit der Echokardiographie nicht direkt gemessen werden. Daher erfordert die Diagnose einer PAH eine Herzkatheteruntersuchung. Mit einem Swan-Ganz-Katheter kann auch das Herzzeitvolumen gemessen werden, das für die Messung der Krankheitsschwere weitaus wichtiger ist als der pulmonal-arterielle Druck.
Der normale pulmonal-arterielle Druck bei einer auf Meereshöhe lebenden Person hat einen Mittelwert von 12-16 mm Hg (1600-2100 Pa). Eine definitive pulmonale Hypertonie liegt vor, wenn der mittlere Druck in Ruhe 25 mm Hg (3300 Pa) übersteigt. Wenn der mittlere Druck in der Lungenarterie bei körperlicher Betätigung über 30 mm Hg (4000 Pa) ansteigt, gilt dies ebenfalls als pulmonale Hypertonie.
Die Diagnose der PAH erfordert das Vorliegen einer pulmonalen Hypertonie mit zwei weiteren Erkrankungen. Der pulmonal-arterielle Verschlussdruck (PAOP oder PCWP) muss weniger als 15 mm Hg (2000 Pa) betragen und der pulmonal-vaskuläre Widerstand (PVR) muss größer als 3 Wood-Einheiten (240 dyn-s-cm-5 oder 2,4 mN-s-cm-5) sein.
Klassifikation
Aktuelle Klassifizierung
Im Jahr 2003 wurde das 3. Weltsymposium über pulmonale arterielle Hypertonie in Venedig einberufen, um die Klassifikation auf der Grundlage des neuen Verständnisses der Krankheitsmechanismen zu modifizieren. Das überarbeitete System, das von dieser Gruppe entwickelt wurde, bildet den aktuellen Rahmen für das Verständnis der pulmonalen Hypertonie.
Das System enthält mehrere Verbesserungen gegenüber dem früheren Evian-Klassifikationssystem von 1998. Die Beschreibungen der Risikofaktoren wurden aktualisiert, und die Klassifikation von angeborenen systemisch-pulmonalen Shunts wurde überarbeitet. Eine neue Klassifikation genetischer Faktoren bei PH wurde empfohlen, aber nicht umgesetzt, da die verfügbaren Daten als unzureichend beurteilt wurden.
Das überarbeitete Klassifizierungssystem von Venedig 2003 lässt sich wie folgt zusammenfassen:
- WHO-Gruppe I - Pulmonal-arterielle Hypertonie (PAH)
- WHO-Gruppe II - Pulmonale Hypertonie in Verbindung mit Linksherzerkrankungen
- WHO-Gruppe III - Pulmonale Hypertonie im Zusammenhang mit Lungenerkrankungen und/oder Hypoxämie
- WHO-Gruppe IV - Pulmonale Hypertonie aufgrund chronischer thrombotischer und/oder embolischer Erkrankungen
- WHO-Gruppe V - Verschiedenes
Frühere Terminologie
Zur Klassifizierung der Erkrankung wurden früher die Begriffe primäre und sekundäre pulmonale Hypertonie (PPH und SPH) verwendet. Dies führte zu der Annahme, dass nur die primäre Erkrankung behandelt werden sollte, während die sekundäre Variante zugunsten der Behandlung nur der zugrunde liegenden Erkrankung ignoriert werden sollte. Tatsächlich sind alle Formen der pulmonal-arteriellen Hypertonie behandelbar. Leider ist dieses Klassifikationssystem immer noch in den Köpfen vieler Ärzte verankert und führt wahrscheinlich dazu, dass vielen Patienten die Behandlung verweigert wird. Diese nihilistische Herangehensweise an die pulmonal-arterielle Hypertonie kann auch zur Unterdiagnose beitragen. Es wird geschätzt, dass es in den USA etwa 100.000 PAH-Patienten gibt, aber nur 15-20.000 wurden bisher diagnostiziert. Viele andere wurden als COPD, Asthma oder kongestive Herzinsuffizienz fehldiagnostiziert.
Der Begriff der primären pulmonalen Hypertonie (PPH) ist in einem Großteil der medizinischen Literatur inzwischen durch den Begriff der idiopathischen pulmonal-arteriellen Hypertonie (IPAH) ersetzt worden. Einige Ärzte verwenden jedoch weiterhin unangemessenerweise die ältere Klassifikation.
Epidemiologie
Die IPAH ist eine seltene Krankheit mit einer Inzidenz von etwa 2-3 pro Million pro Jahr und einer Prävalenz von etwa 15 pro Million. Bei Frauen ist die Wahrscheinlichkeit, an IPAH zu erkranken, fast dreimal so hoch wie bei Männern.
Andere Formen der PAH sind weitaus häufiger anzutreffen. Bei Sklerodermie wird die Inzidenz auf 6 bis 60% aller Patienten geschätzt, bei rheumatoider Arthritis auf bis zu 21%, bei systemischem Lupus erythematodes auf 4 bis 14%, bei portaler Hypertonie auf 2 bis 5%, bei HIV auf etwa 0,5% und bei Sichelzellanämie auf 20 bis 40%.
Diätpillen wie Fen-Phen verursachten eine jährliche Inzidenz von 25-50 pro Million pro Jahr.
Behandlung
Die Behandlung hängt davon ab, ob die PH arteriell, venös, hypoxisch, thromboembolisch oder verschiedenartig ist. Da die pulmonal-venöse Hypertonie gleichbedeutend mit kongestiver Herzinsuffizienz ist, besteht die Behandlung darin, die linksventrikuläre Funktion durch den Einsatz von Diuretika, Betablockern, ACE-Hemmern usw. zu optimieren oder die Mitral- oder Aortenklappe zu reparieren/ersetzen.
Bei PAH gelten Lebensstiländerungen, Digoxin, Diuretika, orale Antikoagulanzien und Sauerstofftherapie als konventionelle Therapie, aber es konnte nie bewiesen werden, dass sie in einer randomisierten, prospektiven Weise vorteilhaft sind.
Hochdosierte Kalziumkanalblocker sind nur bei 5% der IPAH-Patienten nützlich, die mittels Swan-Ganz-Katheter vasoreaktiv sind. Leider wurden Kalziumkanalblocker weitgehend missbräuchlich eingesetzt und vielen Patienten mit nicht-vasoreaktiver PAH verschrieben, was zu übermäßiger Morbidität und Mortalität führte.
Vasoaktive Substanzen
Drei Hauptwege sind an der abnormen Proliferation und Kontraktion der glattmuskulären Zellen der Lungenarterie bei Patienten mit pulmonal-arterieller Hypertonie beteiligt. Diese Signalwege entsprechen wichtigen therapeutischen Zielen bei dieser Erkrankung und spielen eine Rolle bei der Entscheidung, welche der drei Klassen von Medikamenten - Endothelin-Rezeptorantagonisten, Phosphodiesterase-Typ-5-Hemmer und Prostazyklinderivate - eingesetzt werden.
Prostazyklin (Prostaglandin I2) gilt allgemein als die wirksamste Behandlung der PAH. Epoprostenol (synthetisches Prostazyklin, vermarktet als Flolan®) wird über eine kontinuierliche Infusion verabreicht, die einen semi-permanenten zentralvenösen Katheter erfordert. Dieses Verabreichungssystem kann eine Sepsis und Thrombose verursachen. Flolan® ist instabil und muss daher während der Verabreichung auf Eis gelegt werden. Da es eine Halbwertszeit von 3 bis 5 Minuten hat, muss die Infusion kontinuierlich (24/7) erfolgen, und eine Unterbrechung kann tödlich sein. Es wurden daher andere Prostanoide entwickelt. Treprostinil (Remodulin®) kann intravenös oder subkutan verabreicht werden, wobei die subkutane Form sehr schmerzhaft sein kann. Iloprost (Ilomedin®) wird in Europa ebenfalls intravenös verabreicht und hat eine längere Halbwertszeit. Iloprost (vermarktet als Ventavis®) ist die einzige inhalative Form von Prostazyklin, die in den USA und Europa zur Anwendung zugelassen ist. Diese Verabreichungsform hat den Vorteil einer selektiven Ablagerung in der Lunge mit weniger systemischen Nebenwirkungen.
Der duale (ETA und ETB) Endothelin-Rezeptor-Antagonist Bosentan (vermarktet als Tracleer®) wurde 2001 zugelassen. Zwei selektive Endothelin-Rezeptor-Antagonisten (nur ETA) befinden sich in der Endphase der Zulassung: Sitaxsentan und Ambrisentan. Sildenafil, ein selektiver Inhibitor der cGMP-spezifischen Phosphodiesterase Typ 5 (PDE5), wurde 2005 für die Behandlung von PAH zugelassen. Es wird für die Behandlung von PAH als Revatio® vermarktet. Tadalafil (derzeit als Cialis® zur Behandlung der erektilen Dysfunktion vermarktet) befindet sich derzeit in Phase III-Studien. Vasoaktives Darmpeptid durch Inhalation soll 2007 in die klinische Erprobung bei PAH eintreten. PRX-08066 ist ein Serotonin-Antagonist, der derzeit für hypoxische pulmonale Hypertonie entwickelt wird.
Chirurgisch
Die Vorhofseptomie ist ein chirurgischer Eingriff, der eine Kommunikation zwischen dem rechten und linken Vorhof herstellt. Sie entlastet die rechte Seite des Herzens, allerdings auf Kosten eines niedrigeren Sauerstoffgehalts im Blut (Hypoxie). Sie wird am besten in erfahrenen Zentren durchgeführt. Die Lungentransplantation heilt die pulmonal-arterielle Hypertonie, lässt den Patienten jedoch mit den Komplikationen der Transplantation und einer Überlebenszeit von etwa 5 Jahren zurück.
Die pulmonale Thromboendarterektomie (PTE) ist ein chirurgischer Eingriff, der bei chronisch thromboembolischer pulmonaler Hypertonie eingesetzt wird. Es handelt sich dabei um die chirurgische Entfernung eines organisierten Thrombus (Gerinnsel) zusammen mit der Auskleidung der Lungenarterie; es handelt sich um einen großen und sehr schwierigen Eingriff, der gegenwärtig in einigen wenigen ausgewählten Zentren durchgeführt wird. Fallserien zeigen bei den meisten Patienten bemerkenswerte Erfolge.
Eine Behandlung für hypoxische und verschiedene Varianten der pulmonalen Hypertonie ist nicht etabliert. Zurzeit werden jedoch Studien mit mehreren Wirkstoffen an Patienten durchgeführt. Viele Ärzte werden diese Krankheiten mit den gleichen Medikamenten wie bei PAH behandeln, bis bessere Optionen zur Verfügung stehen.
Prognose
Das IPAH-Register der NIH aus den 1980er Jahren zeigte ein unbehandeltes medianes Überleben von 2-3 Jahren ab dem Zeitpunkt der Diagnose, wobei die Todesursache in der Regel rechtsventrikuläre Insuffizienz (cor pulmonale) war. Obwohl diese Zahl häufig zitiert wird, ist sie heute wahrscheinlich irrelevant. Die Ergebnisse haben sich in den letzten zwei Jahrzehnten dramatisch verändert. Dies könnte auf eine neuere medikamentöse Therapie, eine bessere Gesamtversorgung und eine frühere Diagnose zurückzuführen sein (Verzerrung der Vorlaufzeit). Eine kürzlich durchgeführte Ergebnisstudie mit Patienten, die eine Behandlung mit Bosentan (Tracleer®) begonnen hatten, zeigte, dass 86% der Patienten mit 3 Jahren noch lebten. Da inzwischen mehrere Wirkstoffe zur Verfügung stehen, wird zunehmend die Kombinationstherapie eingesetzt. Der Einfluss dieser Wirkstoffe auf das Überleben ist nicht bekannt, da viele von ihnen erst vor kurzem entwickelt wurden. Es wäre nicht unvernünftig zu erwarten, dass sich das mediane Überleben in naher Zukunft auf die letzten 10 Jahre verlängert.
Fragen und Antworten
F: Was ist pulmonale Hypertonie oder PH?
A: Pulmonale Hypertonie oder PH ist ein Zustand, bei dem ein hoher Blutdruck in der Lunge herrscht.
F: Was sind die Symptome der pulmonalen Hypertonie?
A: Zu den Symptomen der pulmonalen Hypertonie gehören Atembeschwerden, Schwindel, Müdigkeit und Ohnmacht.
F: Warum benötigen manche Menschen mit pulmonaler Hypertonie zusätzlichen Sauerstoff?
A: Manche Menschen mit pulmonaler Hypertonie benötigen zusätzlichen Sauerstoff, weil die Erkrankung ihnen das Atmen erschwert.
F: Wann verschlimmern sich die Symptome der pulmonalen Hypertonie?
A: Die Symptome der pulmonalen Hypertonie verschlimmern sich bei sportlicher Betätigung oder schwerer Arbeit.
F: Warum ist pulmonale Hypertonie eine ernste Erkrankung?
A: Pulmonale Hypertonie ist eine ernste Erkrankung, da sie die Pumpleistung des Herzens erschwert und tödlich sein kann.
F: Wie lautet die vollständige Bezeichnung für pulmonale Hypertonie?
A: Der vollständige Name der pulmonalen Hypertonie lautet pulmonale arterielle Hypertonie, auch wenn die meisten Menschen sie als pah, ph oder pha bezeichnen.
F: Was brauchen einige sehr kranke Menschen mit pulmonaler Hypertonie zum Leben?
A: Einige sehr kranke Menschen mit pulmonaler Hypertonie benötigen möglicherweise eine Lungentransplantation oder eine Herz-Lungen-Transplantation, um zu überleben.
Verwandte Artikel
Autor
AlegsaOnline.com Pulmonale Hypertonie (Lungenhochdruck): Ursachen, Symptome & Therapie Leandro Alegsa
URL: https://de.alegsaonline.com/art/80022
Quellen
- ncbi.nlm.nih.gov : PMID 8692238
- ncbi.nlm.nih.gov : PMID 14985486
- ncbi.nlm.nih.gov : PMID 10555089
- ncbi.nlm.nih.gov : PMID 13679525
- ncbi.nlm.nih.gov : PMID 10903931
- ncbi.nlm.nih.gov : PMID 14659797
- ncbi.nlm.nih.gov : PMID 15194171